Encephalocele is a neural tube defect characterized by sac-like protrusions of the brain and the membranes that cover it through openings in the skull. These defects are caused by failure of the neural tube to close completely during fetal development. Encephaloceles cause a groove down the middle of the skull, or between the forehead and nose, or on the back side of the skull. The severity of encephalocele varies, depending on its location.[1]
Encephaloceles are often accompanied by craniofacial abnormalities or other brain malformations. Symptoms may include neurologic problems, hydrocephalus (cerebrospinal fluid accumulated in the brain), spastic quadriplegia (paralysis of the limbs), microcephaly (an abnormally small head), ataxia (uncoordinated muscle movement), developmental delay, vision problems, mental and growth retardation, and seizures.[citation needed]
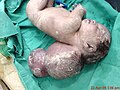
A neonate with a large encephalocele.
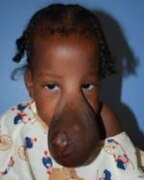
Encephalocele on the head of a two-year-old.
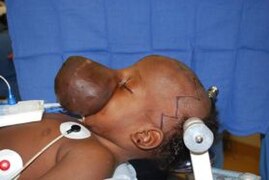
Baby with encephalocele.
Encephalocele.
Neonate with encephalocele
1-year-old with encephalocele and possible microcephaly, 1928
Causes
16-year-old hydrocephalic female patient with occipital encephalocele, 1917
Since its earliest cited case in the 16th century, many generations of scientists have attempted to explain the cause.[2] Little has been revealed in the centuries to follow. Although the exact cause is unknown, encephaloceles are caused by failure of the neural tube to close completely during fetal development. Both environmental and genetic factors have been seen to contribute to the cause of encephaloceles.[3] Some studies have revealed a higher occurrence in female embryos, suggesting a genetic cause.[4] Research has indicated that teratogens (substances known to cause birth defects), trypan blue (a stain used to color dead tissues or cells blue), and arsenic may damage the developing fetus and cause encephaloceles. [citation needed]
Proper levels of folic acid have been shown to help prevent such defects when taken before pregnancy, and early in pregnancy.[citation needed]
Occipital encephaloceles are frequently accompanied by hydrocephalus, as seen in 60-90% of patients.[5]
Diagnosis
Usually encephaloceles are noticeable deformities and are diagnosed immediately after birth, but a small encephalocele in the nasal or forehead region can go undetected. Various physical and mental developmental delays can indicate the presence of encephaloceles.[citation needed]
Classifications
Encephaloceles of the face are generally classified as nasofrontal, nasoethmoidal, or naso-orbital, however, there can be some overlap in the type of encephalocele. They can also appear along any part of the cranial vault, as they result from abnormal closure of cranial bones; the most common location for encephaloceles is the occipital region. If the bulging portion contains only cerebrospinal fluid and the overlying membrane, it may be called a meningocele. If brain tissue is present, it may be referred to as a meningoencephalocele.[6] When the head size or occipitofrontal circumference is smaller than the herniating sac, then it is termed as giant encephalocele.[7]
Separation of the neural and surface ectoderm causes apoptosis in the midline. A disturbance in this separation process at the final closure due to the lack of apoptosis is considered to be a critical aspect of nasofrontal and nasoethmoidal encephalocele.[8]
Prevention
It is recommended that women take a multivitamin with 400 micrograms of folic acid daily to reduce the likelihood of any type of neural tube defects before and during the first 28 days after conception.[9]
Treatment
A series of steps involved in reconstructive surgery of a frontal encephalocele. The same child is in all the images.
1.
2.
3.
4.
5.
6.
Currently, the only effective treatment for encephaloceles is reparative surgery, generally performed during infancy. The extent to which it can be corrected depends on the location and size of the encephaloceles; however, large protrusions can be removed without causing major disability. Surgery repositions the bulging area back into the skull, removes the protrusions, and corrects the deformities, typically relieving pressure that can delay normal brain development. Occasionally, shunts are placed to drain excess cerebrospinal fluid from the brain.[citation needed]
The goals of treatment include:
closure of open skin defects to prevent infection and desiccation of brain tissue
removal of nonfunctional extracranial cerebral tissue with water-tight closure of the dura
total craniofacial reconstruction with particular emphasis on avoiding the long-nose deformity (nasal elongation that results from depression of the cribiform plate and nasal placode). Without proper management, the long-nose deformity can be more obvious after repair.[10]
Recovery
Recovery is difficult to predict prior to surgery, and depends on the type of brain tissue involved and location of the encephaloceles. If surgery is successful, and developmental delays have not occurred, a patient can develop normally. Where neurologic and developmental damage has occurred, the specialists will focus on minimizing both mental and physical disabilities.[citation needed]
In general, when the bulging material consists of primarily cerebrospinal fluid, a complete recovery can occur. When a large amount of brain tissue is present in the encephaloceles, there is a higher chance of perioperative complication.[citation needed]
Epidemiology
Encephaloceles occur rarely, at a rate of one per 5,000 live births worldwide. Encephaloceles of the back of the head are more common in Europe and North America, while encephaloceles on the front of the head more frequently occur in Southeast Asia, Africa, Malaysia, and Russia. Ethnic, genetic, and environmental factors, as well as parental age, can all affect the likelihood of encephaloceles. The condition can occur in families with a family history of spina bifida.[11]
Notable cases
The Facemakers: Operation Smile is a documentary co-produced by the Discovery Channel and BBC 1 in conjunction with century films aired on 21 June 2000.[12] The Facemakers documents the remarkable changes that occurred in the lives of three children as a result of Operation Smile's visit to Davao City in the Philippines in 1999. One child in particular, Abel Gastardo, had a condition too severe to be treated during the time of the mission. Abel had a rare nasofrontal facial encephalocele, an extreme protrusion of brain tissue from the front of his skull. The documentary follows Abel to the United States seven months later to receive corrective surgery. He was brought over by Operation Smile to receive the major surgery in Virginia, at the Children's Hospital of the King's Daughters. The other facial defects within the fifty-minute programme consisted of children with facial cleft and cleft lip and palate which may be associated with encephalocele.[13]
In November 2006, there was an hour-long documentary on the British television network Channel 4 about Facing the World, an organization that helps children with severe facial disfigurements in developing countries. One of the children featured on the documentary was Ney, a Cambodian boy who had a severe form of encephalocele, wherein part of his brain protruded through his face.[citation needed]
On December 4, 2012, Dr. Meara led a cranio-facial surgical team to remove the encephalocele of an infant, Dominic Gundrum, the son of a Wisconsin Court of Appeals judge and his wife. The surgery also closed the baby's skull, repaired a Tessier facial cleft, and brought the baby's facial features together.[14]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.