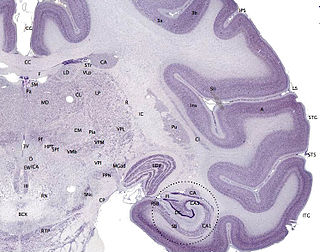
The cerebral cortex, also known as the cerebral mantle, is the outer layer of neural tissue of the cerebrum of the brain in humans and other mammals. The cerebral cortex mostly consists of the six-layered neocortex, with just 10% consisting of the allocortex. It is separated into two cortices, by the longitudinal fissure that divides the cerebrum into the left and right cerebral hemispheres. The two hemispheres are joined beneath the cortex by the corpus callosum. The cerebral cortex is the largest site of neural integration in the central nervous system. It plays a key role in attention, perception, awareness, thought, memory, language, and consciousness. The cerebral cortex is part of the brain responsible for cognition.

Lissencephaly is a set of rare brain disorders whereby the whole or parts of the surface of the brain appear smooth. It is caused by defective neuronal migration during the 12th to 24th weeks of gestation resulting in a lack of development of brain folds (gyri) and grooves (sulci). It is a form of cephalic disorder. Terms such as agyria and pachygyria are used to describe the appearance of the surface of the brain.

Focal cortical dysplasia (FCD) is a congenital abnormality of brain development where the neurons in an area of the brain failed to migrate in the proper formation in utero. Focal means that it is limited to a focal zone in any lobe. Focal cortical dysplasia is a common cause of intractable epilepsy in children and is a frequent cause of epilepsy in adults. There are three types of FCD with subtypes, including type 1a, 1b, 1c, 2a, 2b, 3a, 3b, 3c, and 3d, each with distinct histopathological features. All forms of focal cortical dysplasia lead to disorganization of the normal structure of the cerebral cortex :
Miller–Dieker syndrome, Miller–Dieker lissencephaly syndrome (MDLS), and chromosome 17p13.3 deletion syndrome is a micro deletion syndrome characterized by congenital malformations. Congenital malformations are physical defects detectable in an infant at birth which can involve many different parts of the body including the brain, hearts, lungs, liver, bones, or intestinal tract. MDS is a contiguous gene syndrome – a disorder due to the deletion of multiple gene loci adjacent to one another. The disorder arises from the deletion of part of the small arm of chromosome 17p, leading to partial monosomy. There may be unbalanced translocations, or the presence of a ring chromosome 17.

Polymicrogyria (PMG) is a condition that affects the development of the human brain by multiple small gyri (microgyri) creating excessive folding of the brain leading to an abnormally thick cortex. This abnormality can affect either one region of the brain or multiple regions.
Bilateral frontoparietal polymicrogyria is a genetic disorder with autosomal recessive inheritance that causes a cortical malformation. Our brain has folds in the cortex to increase surface area called gyri and patients with polymicrogyria have an increase number of folds and smaller folds than usual. Polymicrogyria is defined as a cerebral malformation of cortical development in which the normal gyral pattern of the surface of the brain is replaced by an excessive number of small, fused gyri separated by shallow sulci and abnormal cortical lamination. From ongoing research, mutation in GPR56, a member of the adhesion G protein-coupled receptor (GPCR) family, results in BFPP. These mutations are located in different regions of the protein without any evidence of a relationship between the position of the mutation and phenotypic severity. It is also found that GPR56 plays a role in cortical pattering.

In neuroanatomy, a gyrus is a ridge on the cerebral cortex. It is generally surrounded by one or more sulci. Gyri and sulci create the folded appearance of the brain in humans and other mammals.

Gray matter heterotopia is a neurological disorder caused by gray matter being located in an atypical location in the brain.

Neuronal migration protein doublecortin, also known as doublin or lissencephalin-X is a protein that in humans is encoded by the DCX gene.
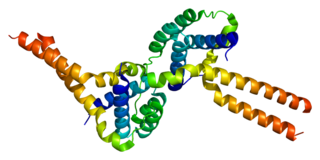
Platelet-activating factor acetylhydrolase IB subunit alpha is an enzyme that in humans is encoded by the PAFAH1B1 gene. The protein is often referred to as Lis1 and plays an important role in regulating the motor protein Dynein.

The ganglionic eminence (GE) is a transitory structure in the development of the nervous system that guides cell and axon migration. It is present in the embryonic and fetal stages of neural development found between the thalamus and caudate nucleus.
Gyrification is the process of forming the characteristic folds of the cerebral cortex.

Neuronal migration disorder (NMD) refers to a heterogenous group of disorders that, it is supposed, share the same etiopathological mechanism: a variable degree of disruption in the migration of neuroblasts during neurogenesis. The neuronal migration disorders are termed cerebral dysgenesis disorders, brain malformations caused by primary alterations during neurogenesis; on the other hand, brain malformations are highly diverse and refer to any insult to the brain during its formation and maturation due to intrinsic or extrinsic causes that ultimately will alter the normal brain anatomy. However, there is some controversy in the terminology because virtually any malformation will involve neuroblast migration, either primarily or secondarily.
Ulegyria is a diagnosis used to describe a specific type of cortical scarring in the deep regions of the sulcus that leads to distortion of the gyri. Ulegyria is identified by its characteristic "mushroom-shaped" gyri, in which scarring causes shrinkage and atrophy in the deep sulcal regions while the surface gyri are spared. This condition is most often caused by hypoxic-ischemic brain injury in the perinatal period. The effects of ulegyria can range in severity, although it is most commonly associated with cerebral palsy, mental retardation and epilepsy. N.C. Bresler was the first to view ulegyria in 1899 and described this abnormal morphology in the brain as “mushroom-gyri." Although ulegyria was first identified in 1899, there is still limited information known or reported about the condition.
The development of the cerebral cortex, known as corticogenesis is the process during which the cerebral cortex of the brain is formed as part of the development of the nervous system of mammals including its development in humans. The cortex is the outer layer of the brain and is composed of up to six layers. Neurons formed in the ventricular zone migrate to their final locations in one of the six layers of the cortex. The process occurs from embryonic day 10 to 17 in mice and between gestational weeks seven to 18 in humans.
Cajal–Retzius cells are a heterogeneous population of morphologically and molecularly distinct reelin-producing cell types in the marginal zone/layer I of the developmental cerebral cortex and in the immature hippocampus of different species and at different times during embryogenesis and postnatal life.

Congenital bilateral perisylvian syndrome (CBPS) is a rare neurological disease characterized by paralysis of certain facial muscles and epileptic seizures.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.

Microlissencephaly (MLIS) is a rare congenital brain disorder that combines severe microcephaly with lissencephaly. Microlissencephaly is a heterogeneous disorder, i.e. it has many different causes and a variable clinical course. Microlissencephaly is a malformation of cortical development (MCD) that occurs due to failure of neuronal migration between the third and fifth month of gestation as well as stem cell population abnormalities. Numerous genes have been found to be associated with microlissencephaly, however, the pathophysiology is still not completely understood.