
Amniocentesis is a medical procedure used primarily in the prenatal diagnosis of genetic conditions. It has other uses such as in the assessment of infection and fetal lung maturity. Prenatal diagnostic testing, which includes amniocentesis, is necessary to conclusively diagnose the majority of genetic disorders, with amniocentesis being the gold-standard procedure after 15 weeks' gestation.
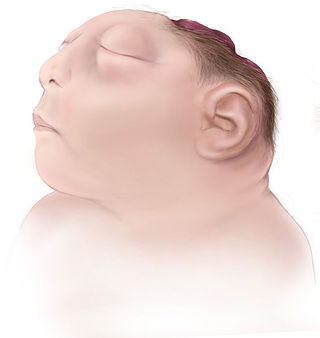
Anencephaly is the absence of a major portion of the brain, skull, and scalp that occurs during embryonic development. It is a cephalic disorder that results from a neural tube defect that occurs when the rostral (head) end of the neural tube fails to close, usually between the 23rd and 26th day following conception. Strictly speaking, the Greek term translates as "without a brain", but it is accepted that children born with this disorder usually only lack a telencephalon, the largest part of the brain consisting mainly of the cerebral hemispheres, including the neocortex, which is responsible for cognition. The remaining structure is usually covered only by a thin layer of membrane—skin, bone, meninges, etc., are all lacking. With very few exceptions, infants with this disorder do not survive longer than a few hours or days after birth.

Holoprosencephaly (HPE) is a cephalic disorder in which the prosencephalon fails to develop into two hemispheres, typically occurring between the 18th and 28th day of gestation. Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species.
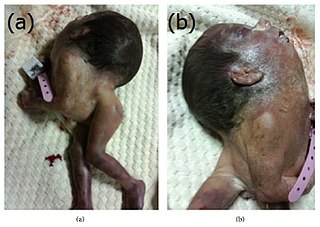
Iniencephaly is a rare type of cephalic disorder characterised by three common characteristics: a defect to the occipital bone, spina bifida of the cervical vertebrae and retroflexion of the head on the cervical spine. Stillbirth is the most common outcome, with a few rare examples of live birth, after which death invariably occurs within a short time.

Prenatal testing is a tool that can be used to detect some birth defects at various stages prior to birth. Prenatal testing consists of prenatal screening and prenatal diagnosis, which are aspects of prenatal care that focus on detecting problems with the pregnancy as early as possible. These may be anatomic and physiologic problems with the health of the zygote, embryo, or fetus, either before gestation even starts or as early in gestation as practicable. Screening can detect problems such as neural tube defects, chromosome abnormalities, and gene mutations that would lead to genetic disorders and birth defects, such as spina bifida, cleft palate, Down syndrome, trisomy 18, Tay–Sachs disease, sickle cell anemia, thalassemia, cystic fibrosis, muscular dystrophy, and fragile X syndrome. Some tests are designed to discover problems which primarily affect the health of the mother, such as PAPP-A to detect pre-eclampsia or glucose tolerance tests to diagnose gestational diabetes. Screening can also detect anatomical defects such as hydrocephalus, anencephaly, heart defects, and amniotic band syndrome.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Because the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects. The syndrome is caused by a mutation in a gene on chromosome 10 that controls the body's production of fibroblast growth factor receptor 2 (FGFR2).

Smith–Lemli–Opitz syndrome is an inborn error of cholesterol synthesis. It is an autosomal recessive, multiple malformation syndrome caused by a mutation in the enzyme 7-Dehydrocholesterol reductase encoded by the DHCR7 gene. It causes a broad spectrum of effects, ranging from mild intellectual disability and behavioural problems to lethal malformations.

Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis. This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly). Individuals with more severe cases of SCS may have mild to moderate intellectual or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention. Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.

Carpenter syndrome, also called acrocephalopolysyndactyly type II, is an extremely rare autosomal recessive congenital disorder characterized by craniofacial malformations, obesity, syndactyly, and polydactyly. Acrocephalopolysyndactyly is a variation of acrocephalosyndactyly that presents with polydactyly.
Acalvaria is a rare malformation consisting of the absence of the calvarial bones, dura mater and associated muscles in the presence of a normal skull base and normal facial bones. The central nervous system is usually unaffected. The presumed pathogenesis of acalvaria is the faulty migration of the membranous neurocranium with normal placement of the embryonic ectoderm, resulting in the absence of the calvaria, but with an intact layer of skin over the brain parenchyma. In other words, instead of having a skull cap protecting the brain, there is only skin covering it. The size of the area that is missing the skull cap can vary from case to case. In extreme cases, the entire top part of the cranium that is dome-shaped may be absent.

Simpson–Golabi–Behmel syndrome (SGBS) is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.
An osteochondrodysplasia, or skeletal dysplasia, is a disorder of the development of bone and cartilage. Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. These disorders lead to disproportionate short stature and bone abnormalities, particularly in the arms, legs, and spine. Skeletal dysplasia can result in marked functional limitation and even mortality.
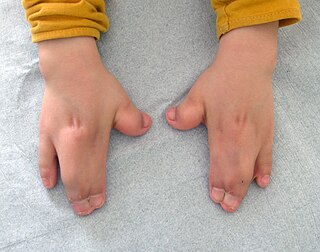
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.

Campomelic dysplasia (CMD) is a genetic disorder characterized by bowing of the long bones and many other skeletal and extraskeletal features. It can be lethal in the neonatal period due to respiratory insufficiency, but the severity of the disease is variable, and a significant proportion of patients survive into adulthood. The name is derived from the Greek roots campo, meaning bent, and melia, meaning limb. An unusual aspect of the disease is that up to two-thirds of affected 46,XY genotypic males display a range of disorders of sexual development (DSD) and genital ambiguities or may even develop as normal phenotypic females as in complete 46 XY sex reversal. An atypical form of the disease with absence of bowed limbs is called, prosaically, acampomelic campomelic dysplasia (ACD) and is found in about 10% of patients, particularly those surviving the neonatal period.
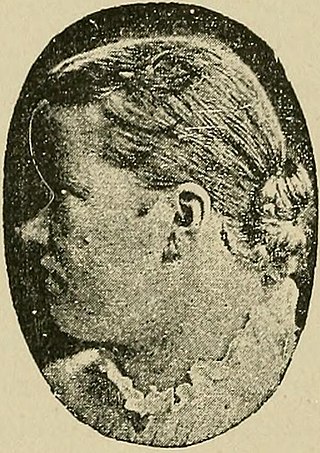
Muenke syndrome, also known as FGFR3-related craniosynostosis, is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
3-M syndrome or 3M3 is a rare hereditary disorder characterized by severe growth retardation, facial dysmorphia, and skeletal abnormalities. The name 3-M is derived from the initials of the three researchers who first identified it: Miller, McKusick, and Malvaux and report their findings in the medical literature in 1972. Mutations in any one of the following three genes: CUL7, OBSL1, and CCDC8 are responsible for the occurrence of this disorder. It is inherited through an autosomal recessive pattern and considered very rare, so far less than 100 cases worldwide have been identified. Diagnosis is based on the presence of clinical features. Genetic testing can confirm the diagnosis and identify the specific gene involved. Treatment is aimed at addressing the growth and skeletal problems and may include surgical bone lengthening, adaptive aids, and physical therapy. An endocrinologist may assist with growth hormone replacement and appropriate evaluations during puberty.
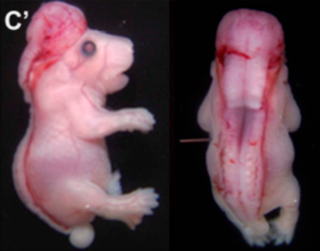
Rachischisis is a developmental birth defect involving the neural tube. This anomaly occurs in utero, when the posterior neuropore of the neural tube fails to close by the 27th intrauterine day. As a consequence the vertebrae overlying the open portion of the spinal cord do not fully form and remain unfused and open, leaving the spinal cord exposed. Patients with rachischisis have motor and sensory deficits, chronic infections, and disturbances in bladder function. This defect often occurs with anencephaly.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).

