Related Research Articles
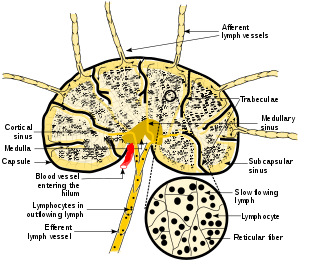
A lymph node, or lymph gland, is a kidney-shaped organ of the lymphatic system and the adaptive immune system. A large number of lymph nodes are linked throughout the body by the lymphatic vessels. They are major sites of lymphocytes that include B and T cells. Lymph nodes are important for the proper functioning of the immune system, acting as filters for foreign particles including cancer cells, but have no detoxification function.
Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.

Lymphadenopathy or adenopathy is a disease of the lymph nodes, in which they are abnormal in size or consistency. Lymphadenopathy of an inflammatory type is lymphadenitis, producing swollen or enlarged lymph nodes. In clinical practice, the distinction between lymphadenopathy and lymphadenitis is rarely made and the words are usually treated as synonymous. Inflammation of the lymphatic vessels is known as lymphangitis. Infectious lymphadenitis affecting lymph nodes in the neck is often called scrofula.

Anaplastic large cell lymphoma (ALCL) refers to a group of non-Hodgkin lymphomas in which aberrant T cells proliferate uncontrollably. Considered as a single entity, ALCL is the most common type of peripheral lymphoma and represents ~10% of all peripheral lymphomas in children. The incidence of ALCL is estimated to be 0.25 cases per 100,000 people in the United States of America. There are four distinct types of anaplastic large cell lymphomas that on microscopic examination share certain key histopathological features and tumor marker proteins. However, the four types have very different clinical presentations, gene abnormalities, prognoses, and/or treatments.
Follicular lymphoma (FL) is a cancer that involves certain types of white blood cells known as lymphocytes. The cancer originates from the uncontrolled division of specific types of B-cells known as centrocytes and centroblasts. These cells normally occupy the follicles in the germinal centers of lymphoid tissues such as lymph nodes. The cancerous cells in FL typically form follicular or follicle-like structures in the tissues they invade. These structures are usually the dominant histological feature of this cancer.
Splenic marginal zone lymphoma (SMZL) is a type of cancer made up of B-cells that replace the normal architecture of the white pulp of the spleen. The neoplastic cells are both small lymphocytes and larger, transformed lymphoblasts, and they invade the mantle zone of splenic follicles and erode the marginal zone, ultimately invading the red pulp of the spleen. Frequently, the bone marrow and splenic hilar lymph nodes are involved along with the peripheral blood. The neoplastic cells circulating in the peripheral blood are termed villous lymphocytes due to their characteristic appearance.
Diffuse large B-cell lymphoma (DLBCL) is a cancer of B cells, a type of lymphocyte that is responsible for producing antibodies. It is the most common form of non-Hodgkin lymphoma among adults, with an annual incidence of 7–8 cases per 100,000 people per year in the US and UK. This cancer occurs primarily in older individuals, with a median age of diagnosis at ~70 years, although it can occur in young adults and, in rare cases, children. DLBCL can arise in virtually any part of the body and, depending on various factors, is often a very aggressive malignancy. The first sign of this illness is typically the observation of a rapidly growing mass or tissue infiltration that is sometimes associated with systemic B symptoms, e.g. fever, weight loss, and night sweats.
Angioimmunoblastic T-cell lymphoma is a mature T-cell lymphoma of blood or lymph vessel immunoblasts characterized by a polymorphous lymph node infiltrate showing a marked increase in follicular dendritic cells (FDCs) and high endothelial venules (HEVs) and systemic involvement.
Lymphoid hyperplasia is the rapid proliferation of normal lymphocytic cells that resemble lymph tissue which may occur with bacterial or viral infections. The growth is termed hyperplasia which may result in enlargement of various tissue including an organ, or cause a cutaneous lesion.
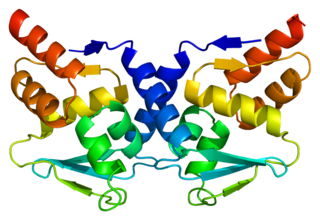
Bcl-6 is a protein that in humans is encoded by the BCL6 gene. BCL6 is a master transcription factor for regulation of T follicular helper cells proliferation. BCL6 has three evolutionary conserved structural domains. The interaction of these domains with corepressors allows for germinal center development and leads to B cell proliferation.

Interferon regulatory factor 4 (IRF4) also known as MUM1 is a protein that in humans is encoded by the IRF4 gene, located at 6p25-p23. IRF4 functions as a key regulatory transcription factor in the development of human immune cells. The expression of IRF4 is essential for the differentiation of T lymphocytes and B lymphocytes as well as certain myeloid cells.

Enteropathy-associated T-cell lymphoma (EATL), previously termed enteropathy-associated T-cell lymphoma, type I and at one time termed enteropathy-type T-cell lymphoma (ETTL), is a complication of coeliac disease in which a malignant T-cell lymphoma develops in areas of the small intestine affected by the disease's intense inflammation. While a relatively rare disease, it is the most common type of primary gastrointestinal T-cell lymphoma.
Nodal marginal zone B cell lymphoma (NMZL) is an uncommon form of marginal-zone lymphoma that can produce colonization of the follicles in the lymph node. It is a form of low grade lymphoma with similar incidence in men and women and a mean age of 61 years. It is often associated with Sjogren syndrome. It shows interfollicular infiltrate of monocytoid, centrocyte-like B cells that are 2–3× larger than small lymphocytes with partial/total effacement of lymph node architecture.

Marginal zone B-cell lymphomas, also known as marginal zone lymphomas (MZLs), are a heterogeneous group of lymphomas that derive from the malignant transformation of marginal zone B-cells. Marginal zone B cells are innate lymphoid cells that normally function by rapidly mounting IgM antibody immune responses to antigens such as those presented by infectious agents and damaged tissues. They are lymphocytes of the B-cell line that originate and mature in secondary lymphoid follicles and then move to the marginal zones of mucosa-associated lymphoid tissue, the spleen, or lymph nodes. Mucosa-associated lymphoid tissue is a diffuse system of small concentrations of lymphoid tissue found in various submucosal membrane sites of the body such as the gastrointestinal tract, mouth, nasal cavity, pharynx, thyroid gland, breast, lung, salivary glands, eye, skin and the human spleen.

Follicular hyperplasia (FH) is a type of lymphoid hyperplasia and is classified as a lymphadenopathy, which means a disease of the lymph nodes. It is caused by a stimulation of the B cell compartment and by abnormal cell growth of secondary follicles. This typically occurs in the cortex without disrupting the lymph node capsule. The follicles are pathologically polymorphous, are often contrasting and varying in size and shape. Follicular hyperplasia is distinguished from follicular lymphoma in its polyclonality and lack of bcl-2 protein expression, whereas follicular lymphoma is monoclonal, and expresses bcl-2.
Epstein–Barr virus–associated lymphoproliferative diseases are a group of disorders in which one or more types of lymphoid cells, i.e. B cells, T cells, NK cells, and histiocytic-dendritic cells, are infected with the Epstein–Barr virus (EBV). This causes the infected cells to divide excessively, and is associated with the development of various non-cancerous, pre-cancerous, and cancerous lymphoproliferative disorders (LPDs). These LPDs include the well-known disorder occurring during the initial infection with the EBV, infectious mononucleosis, and the large number of subsequent disorders that may occur thereafter. The virus is usually involved in the development and/or progression of these LPDs although in some cases it may be an "innocent" bystander, i.e. present in, but not contributing to, the disease.
In situ lymphoid neoplasia is a precancerous condition newly classified by the World Health Organization in 2016. The Organization recognized two subtypes of ISLN: in situ follicular neoplasia (ISFN) and in situ mantle cell neoplasia (ISMCL). ISFN and ISMCL are pathological accumulations of lymphocytes in the germinal centers and mantle zones, respectively, of the follicles that populate lymphoid organs such as lymph nodes. These lymphocytes are monoclonal B-cells that may develop into follicular (FL) and mantle cell (MCL) lymphomas, respectively.
Duodenal-type follicular lymphoma (DFL) is a form of lymphoma in which certain lymphocyte types, the B-cell-derived centrocytes and centroblasts, form lymph node follicle-like structures principally in the duodenum and other parts of the small intestine. It is an indolent disease which on rare occasions progresses to a more aggressive lymphoma that spreads beyond these originally involved sites.
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a malignancy of B cells. B-cells are lymphocytes that normally function in the humoral immunity component of the adaptive immune system by secreting antibodies that, for example, bind to and neutralize invasive pathogens. Among the various forms of B-cell lymphomas, THRLBCL is a rarely occurring subtype of the diffuse large B-cell lymphomas (DLBCL). DLBCL are a large group of lymphomas that account for ~25% of all non-Hodgkin lymphomas worldwide. THRLBCL is distinguished from the other DLBCL subtypes by the predominance of non-malignant T-cell lymphocytes and histiocytes over malignant B-cells in its tumors and tissue infiltrates.
T-Cell Lymphoblastic Leukemia (T-ALL) is a type of acute lymphoblastic leukemia with aggressive malignant neoplasm of the bone marrow. Acute Lymphoblastic Leukemia (ALL) is a condition where immature white blood cells accumulate in the bone marrow, subsequently crowding out normal white blood cells and create build-up in the liver, spleen, and lymph nodes. The two most common types of ALL are B-Lymphocytes and T-Lymphocytes, where the first protects the body against viruses and bacteria through antibody production which can directly destroy target cells or trigger others to do so, whilst the latter directly destroy bacteria or cells infected with viruses. Approximately 20% of all ALL patients are categorized specifically to suffer from T-ALL and it is seen to be more prevalent in the adult population in comparison to children, with incidences shown to diminish with age. Amongst T-ALL cases in the pediatric population, a median onset of age 9 has been identified and the disease is particularly prominent amongst adolescents. The disease stems from cytogenic and molecular abnormalities, resulting in disruption of developmental pathways controlling thymocyte development, tumor suppressor development, and alterations in control of cell growth and proliferation. Distinct from adult T-Cell Leukemia where T-Cell Lymphotropic Virus Type I causes malignant maturation of T-cells, T-ALL is a precursor for lymphoid neoplasm. Its clinical presentation most commonly includes infiltration of the central nervous system (CNS), and further identifies mediastinal mass presence originating from the thymus, along with extramedullary involvement of multiple organs including the lymph node as a result of hyperleukocytosis.
References
- ↑ Quintanilla-Martinez L, Sander B, Chan JK, Xerri L, Ott G, Campo E, Swerdlow SH (February 2016). "Indolent lymphomas in the pediatric population: follicular lymphoma, IRF4/MUM1+ lymphoma, nodal marginal zone lymphoma and chronic lymphocytic leukemia". Virchows Archiv. 468 (2): 141–57. doi:10.1007/s00428-015-1855-z. PMID 26416032. S2CID 531366.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Koo M, Ohgami RS (May 2017). "Pediatric-type Follicular Lymphoma and Pediatric Nodal Marginal Zone Lymphoma: Recent Clinical, Morphologic, Immunophenotypic, and Genetic Insights". Advances in Anatomic Pathology. 24 (3): 128–135. doi:10.1097/PAP.0000000000000144. PMID 28277421.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Woessmann W, Quintanilla-Martinez L (June 2019). "Rare mature B-cell lymphomas in children and adolescents". Hematological Oncology. 37 Suppl 1: 53–61. doi: 10.1002/hon.2585 . PMID 31187530.
- 1 2 3 4 5 Lynch RC, Gratzinger D, Advani RH (July 2017). "Clinical Impact of the 2016 Update to the WHO Lymphoma Classification". Current Treatment Options in Oncology. 18 (7): 45. doi:10.1007/s11864-017-0483-z. PMID 28670664. S2CID 4415738.
- ↑ Du XY, Huang R, Cao L, Wu W, Wang Z, Zhu HY, Wang L, Fan L, Xu W, Li JY (May 2019). "[Clinical observation of five pediatric-type follicular lymphoma in adult]". Zhonghua Xue Ye Xue Za Zhi = Zhonghua Xueyexue Zazhi (in Chinese). 40 (5): 393–397. doi:10.3760/cma.j.issn.0253-2727.2019.05.009. PMC 7342233 . PMID 31207704.
- ↑ Yamaguchi J, Kato S, Iwata E, Aoki K, Kabeya R, Natsume A, Wakabayashi T (July 2018). "Pediatric-Type Follicular Lymphoma in the Dura: A Case Report and Literature Review". World Neurosurgery. 115: 176–180. doi:10.1016/j.wneu.2018.04.053. PMID 29678710. S2CID 5029006.
- 1 2 Louissaint A, Schafernak KT, Geyer JT, Kovach AE, Ghandi M, Gratzinger D, Roth CG, Paxton CN, Kim S, Namgyal C, Morin R, Morgan EA, Neuberg DS, South ST, Harris MH, Hasserjian RP, Hochberg EP, Garraway LA, Harris NL, Weinstock DM (August 2016). "Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations". Blood. 128 (8): 1093–100. doi:10.1182/blood-2015-12-682591. PMC 5000844 . PMID 27325104.
- 1 2 Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (May 2016). "The 2016 revision of the World Health Organization classification of lymphoid neoplasms". Blood. 127 (20): 2375–90. doi:10.1182/blood-2016-01-643569. PMC 4874220 . PMID 26980727.
- ↑ "IRF4 interferon regulatory factor 4 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov.
- ↑ "IGH immunoglobulin heavy locus [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov.
- 1 2 3 Liu Q, Salaverria I, Pittaluga S, Jegalian AG, Xi L, Siebert R, Raffeld M, Hewitt SM, Jaffe ES (March 2013). "Follicular lymphomas in children and young adults: a comparison of the pediatric variant with usual follicular lymphoma". The American Journal of Surgical Pathology. 37 (3): 333–43. doi:10.1097/PAS.0b013e31826b9b57. PMC 3566339 . PMID 23108024.
- ↑ EntrezGene 7128
- ↑ EntrezGene 8764
- ↑ Shukla V, Lu R (August 2014). "IRF4 and IRF8: Governing the virtues of B Lymphocytes". Frontiers in Biology. 9 (4): 269–282. doi:10.1007/s11515-014-1318-y. PMC 4261187 . PMID 25506356.
- ↑ "IRF8 interferon regulatory factor 8 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov.
- ↑ "MAP2K1 mitogen-activated protein kinase kinase 1 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov.
- ↑ Lovisa F, Binatti A, Coppe A, Primerano S, Carraro E, Pillon M, Pizzi M, Guzzardo V, Buffardi S, Porta F, Farruggia P, De Santis R, Bulian P, Basso G, Lazzari E, d'Amore ES, Bortoluzzi S, Mussolin L (February 2019). "A high definition picture of key genes and pathways mutated in pediatric follicular lymphoma". Haematologica. 104 (9): e406–e409. doi:10.3324/haematol.2018.211631. PMC 6717562 . PMID 30819919.