
Tetrahedrane is a hypothetical platonic hydrocarbon with chemical formula C4H4 and a tetrahedral structure. The molecule would be subject to considerable angle strain and has not been synthesized as of 2021. However, a number of derivatives have been prepared. In a more general sense, the term tetrahedranes is used to describe a class of molecules and ions with related structure, e.g. white phosphorus.

A silabenzene is a heteroaromatic compound containing one or more silicon atoms instead of carbon atoms in benzene. A single substitution gives silabenzene proper; additional substitutions give a disilabenzene, trisilabenzene, etc.
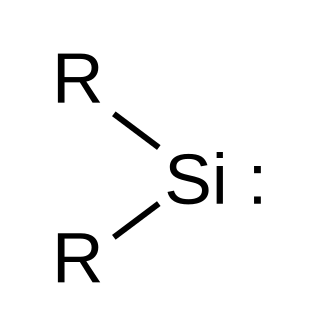
Silylene is a chemical compound with the formula SiH2. It is the silicon analog of methylene, the simplest carbene. Silylene is a stable molecule as a gas but rapidly reacts in a bimolecular manner when condensed. Unlike carbenes, which can exist in the singlet or triplet state, silylene (and all of its derivatives) are singlets.

Organotin chemistry is the scientific study of the synthesis and properties of organotin compounds or stannanes, which are organometallic compounds containing tin–carbon bonds. The first organotin compound was diethyltin diiodide, discovered by Edward Frankland in 1849. The area grew rapidly in the 1900s, especially after the discovery of the Grignard reagents, which are useful for producing Sn–C bonds. The area remains rich with many applications in industry and continuing activity in the research laboratory.
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.
Diphosphene is a type of organophosphorus compound that has a phosphorus–phosphorus double bond, denoted by R-P=P-R'. These compounds are not common but are of theoretical interest. Normally, compounds with the empirical formula RP exist as rings. However, like other multiple bonds between heavy main-group elements, P=P double bonds can be stabilized by a large steric hindrance from the substitutions. The first isolated diphosphene bis(2,4,6-tri-tert-butylphenyl)diphosphene was exemplified by Masaaki Yoshifuji and his coworkers in 1981, in which diphosphene is stabilized by two bulky phenyl group.

In chemistry, a phosphaalkyne is an organophosphorus compound containing a triple bond between phosphorus and carbon with the general formula R-C≡P. Phosphaalkynes are the heavier congeners of nitriles, though, due to the similar electronegativities of phosphorus and carbon, possess reactivity patterns reminiscent of alkynes. Due to their high reactivity, phosphaalkynes are not found naturally on earth, but the simplest phosphaalkyne, phosphaethyne (H-C≡P) has been observed in the interstellar medium.
In chemistry, the double bond rule states that elements with a principal quantum number (n) greater than 2 for their valence electrons (period 3 elements and higher) tend not to form multiple bonds (e.g. double bonds and triple bonds). The double bonds, when they exist, are often weak due to poor orbital overlap between the n>2 orbitals of the two atoms. Although such compounds are not intrinsically unstable, they instead tend to polymerize. An example is the rapid polymerization that occurs upon condensation of disulfur, the heavy analogue of O2. Numerous violations to the rule exist.
Carbene analogs in chemistry are carbenes with the carbon atom replaced by another chemical element. Just as regular carbenes they appear in chemical reactions as reactive intermediates and with special precautions they can be stabilized and isolated as chemical compounds. Carbenes have some practical utility in organic synthesis but carbene analogs are mostly laboratory curiosities only investigated in academia. Carbene analogs are known for elements of group 13, group 14, group 15 and group 16.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.

Phosphinidenes are low-valent phosphorus compounds analogous to carbenes and nitrenes, having the general structure RP. The "free" form of these compounds is conventionally described as having a singly-coordinated phosphorus atom containing only 6 electrons in its valence level. Most phosphinidenes are highly reactive and short-lived, thereby complicating empirical studies on their chemical properties. In the last few decades, several strategies have been employed to stabilize phosphinidenes, and researchers have developed a number of reagents and systems that can generate and transfer phosphinidenes as reactive intermediates in the synthesis of various organophosphorus compounds.

tert-Butylphosphaacetylene is an organophosphorus compound. Abbreviated t-BuCP, it was the first example of an isolable phosphaalkyne. Prior to its synthesis, the double bond rule had suggested that elements of Period 3 and higher were unable to form double or triple bonds with lighter main group elements because of weak orbital overlap. The synthesis of t-BuCP discredited much of the double bond rule and opened new studies into the formation of unsaturated phosphorus compounds.
Hydrophosphination is the insertion of a carbon-carbon multiple bond into a phosphorus-hydrogen bond forming a new phosphorus-carbon bond. Like other hydrofunctionalizations, the rate and regiochemistry of the insertion reaction is influenced by the catalyst. Catalysts take many forms, but most prevalent are bases and free-radical initiators. Most hydrophosphinations involve reactions of phosphine (PH3).
Digermynes are a class of compounds that are regarded as the heavier digermanium analogues of alkynes. The parent member of this entire class is HGeGeH, which has only been characterized computationally, but has revealed key features of the whole class. Because of the large interatomic repulsion between two Ge atoms, only kinetically stabilized digermyne molecules can be synthesized and characterized by utilizing bulky protecting groups and appropriate synthetic methods, for example, reductive coupling of germanium(II) halides.

Diphosphagermylenes are a class of compounds containing a divalent germanium atom bound to two phosphorus atoms. While these compounds resemble diamidocarbenes, such as N-heterocyclic carbenes (NHC), diphosphagermylenes display bonding characteristics distinct from those of diamidocarbenes. In contrast to NHC compounds, in which there is effective N-C p(π)-p(π) overlap between the lone pairs of planar nitrogens and an empty p-orbital of a carbene, systems containing P-Ge p(π)-p(π) overlap are rare. Until 2014, the geometry of phosphorus atoms in all previously reported diphosphatetrylenes are pyramidal, with minimal P-Ge p(π)-p(π) interaction. It has been suggested that the lack of p(π)-p(π) in Ge-P bonds is due to the high energetic barrier associated with achieving a planar configuration at phosphorus, which would allow for efficient p(π)-p(π) overlap between the phosphorus lone pair and the empty P orbital of Ge. The resulting lack of π stabilization contributes to the difficulty associated with isolating diphosphagermylene and the Ge-P double bonds. However, utilization of sterically encumbering phosphorus centers has allowed for the isolation of diphosphagermylenes with a planar phosphorus center with a significant P-Ge p(π)-p(π) interaction.

Trisilaallene is a subclass of silene derivatives where a central silicon atom forms double bonds with each of two terminal silicon atoms, with the generic formula R2Si=Si=SiR2. Trisilaallene is a silicon-based analog of an allene, but their chemical properties are markedly different.

An N-Heterocyclic silylene (NHSi) is an uncharged heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis. Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.

Plumbylenes (or plumbylidenes) are divalent organolead(II) analogues of carbenes, with the general chemical formula, R2Pb, where R denotes a substituent. Plumbylenes possess 6 electrons in their valence shell, and are considered open shell species.
1-Phosphaallenes is are allenes in which the first carbon atom is replaced by phosphorus, resulting in the structure: -P=C=C<.
Negative hyperconjugation is a theorized phenomenon in organosilicon compounds, in which hyperconjugation stabilizes or destabilizes certain accumulations of positive charge. The phenomenon explains corresponding peculiarities in the stereochemistry and rate of hydrolysis.
















