
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosome abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.

Metachondromatosis is an autosomal dominant, incompletely penetrant genetic disease affecting the growth of bones, leading to exostoses primarily in the hands and feet as well as enchondromas of long bone metaphyses and iliac crests. This syndrome affects mainly tubular bones, though it can also involve the vertebrae, small joints, and flat bones. The disease is thought to affect exon 4 of the PTPN11 gene. Metachondromatosis is believed to be caused by an 11 base pair deletion resulting in a frameshift and nonsense mutation. The disease was discovered and named in 1971 by Pierre Maroteaux, a French physician, when he observed two families with skeletal radiologic features with exostoses and Ollier disease. The observation of one family with five affected people led to the identification of the disease as autosomal dominant. There have been less than 40 cases of the disease reported to date.
Abruzzo–Erickson syndrome is an extremely rare disorder characterized by deafness, protruding ears, coloboma, a cleft palate or palatal rugosity, radial synostosis, and short stature. It was first characterized by Abruzzo and Erickson in 1977 as a CHARGE like syndrome as variably expressed among a family of two brothers, their mother, and their maternal uncle. Members of this family exhibited many of the CHARGE symptoms, but notably did not have choanal atresia and the brothers experienced typical genital development. Due to the recent discovery of this disorder, its etiology is not fully known but it is understood that it arises from mutations on the TBX22 gene on the X-chromosome. The disorder is inherited in an X-linked recessive manner. There is currently no known cure but its symptoms can be treated.
Acromicric dysplasia is an extremely rare inherited disorder characterized by abnormally short hands and feet, growth retardation and delayed bone maturation leading to short stature. Most cases have occurred randomly for no apparent reason (sporadically). However, autosomal dominant inheritance has not been ruled out.
An osteochondrodysplasia, or skeletal dysplasia, is a disorder of the development of bone and cartilage. Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. These disorders lead to disproportionate short stature and bone abnormalities, particularly in the arms, legs, and spine. Skeletal dysplasia can result in marked functional limitation and even mortality.
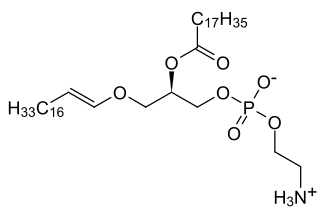
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by abnormally short arms and legs (rhizomelia), seizures, recurrent respiratory tract infections and congenital cataracts.

Pseudoachondroplasia is an inherited disorder of bone growth. It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.
Acrodysostosis is a rare congenital malformation syndrome which involves shortening of the interphalangeal joints of the hands and feet, intellectual disability in approximately 90% of affected children, and peculiar facies. Other common abnormalities include short head, small broad upturned nose with flat nasal bridge, protruding jaw, increased bone age, intrauterine growth retardation, juvenile arthritis and short stature. Further abnormalities of the skin, genitals, teeth, and skeleton may occur.
Danon disease is a metabolic disorder. Danon disease is an X-linked lysosomal and glycogen storage disorder associated with hypertrophic cardiomyopathy, skeletal muscle weakness, and intellectual disability. It is inherited in an X-linked dominant pattern.
Acromesomelic dysplasia is a rare skeletal disorder that causes abnormal bone and cartilage development, leading to shortening of the forearms, lower legs, hands, feet, fingers, and toes. Five different genetic mutations have been implicated in the disorder. Treatment is individualized but is generally aimed at palliating symptoms, for example, treatment of kyphosis and lumbar hyperlordosis.
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) is a very rare genetic disorder. This disorder is one that affects bone growth and is characterized by skeletal, brain, and skin abnormalities. Those affected by the disorder are severely short in height and commonly possess shorter arms and legs. In addition, the bones of the legs are often bowed and the affected have smaller chests with shorter rib bones, along with curved collarbones. Other symptoms of the disorder include broad fingers and extra folds of skin on the arms and legs. Developmentally, many individuals who suffer from the disorder show a higher level in delays and disability. Seizures are also common due to structural abnormalities of the brain. Those affected may also suffer with apnea, the slowing or loss of breath for short periods of time.

Rhizomelic dysplasia, scoliosis, and retinitis pigmentosa is a very rare genetic disorder which is characterized by ocular/visual, dental and osseous anomalies. Only 2 cases have been described in medical literature.

Spondyloepimetaphyseal dysplasia-short limb-abnormal calcification syndrome is a rare genetic disorder which is characterized by osseous anomalies resulting in short stature and other afflictions.
Schneckenbecken dysplasia is a rare pre-natally fatal hereditary autosomal recessive condition which affects the bones and pre-natal growth.
Spondylometaphyseal dysplasia with cone-rod dystrophy is a rare genetic disorder characterized by spondylometaphyseal dysplasia, neonatal growth delays, and cone-rod dystrophy-associated progressive vision loss. Only 18 patients from families in the United States, the United Kingdom, Japan, and Brazil have been described to date. This condition is caused by autosomal recessive mutations in the PCYT1A gene, located in chromosome 3.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.

Calvarial doughnut lesions-bone fragility syndrome, also known as familial calvarial doughnut lesions, is a rare autosomal dominant genetic disorder characterized by mild to moderate fragility of the bones accompanied with calvarial doughnut lesions.
SOFT syndrome, also known for the name its acronym originates from: Short stature-onychodysplasia-facial dysmorphism-hypotrichosis syndrome, is a rare genetic disorder characterized by the presence of short stature, underdeveloped nails, facial dysmorphisms, and hair sparcity across the body. It is caused by homozygous, autosomal recessive mutations in the POC1A gene, located in the short arm of chromosome 3. Fewer than 15 cases have been described in the medical literature.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
