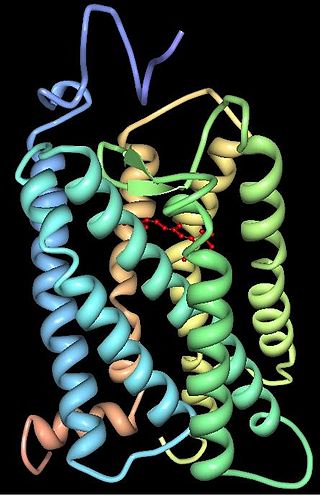
Rhodopsin, also known as visual purple, is a protein encoded by the RHO gene and a G-protein-coupled receptor (GPCR). It is the opsin of the rod cells in the retina and a light-sensitive receptor protein that triggers visual phototransduction in rods. Rhodopsin mediates dim light vision and thus is extremely sensitive to light. When rhodopsin is exposed to light, it immediately photobleaches. In humans, it is regenerated fully in about 30 minutes, after which the rods are more sensitive. Defects in the rhodopsin gene cause eye diseases such as retinitis pigmentosa and congenital stationary night blindness.

Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.
Rhodopsin kinase is a serine/threonine-specific protein kinase involved in phototransduction. This enzyme catalyses the following chemical reaction:

The photoreceptor cell-specific nuclear receptor (PNR), also known as NR2E3, is a protein that in humans is encoded by the NR2E3 gene. PNR is a member of the nuclear receptor super family of intracellular transcription factors.

Peropsin, a visual pigment-like receptor, is a protein that in humans is encoded by the RRH gene. It belongs like other animal opsins to the G protein-coupled receptors. Even so, the first peropsins were already discovered in mice and humans in 1997, not much is known about them.

RPE-retinal G protein-coupled receptor also known as RGR-opsin is a protein that in humans is encoded by the RGR gene. RGR-opsin is a member of the rhodopsin-like receptor subfamily of GPCR. Like other opsins which bind retinaldehyde, it contains a conserved lysine residue in the seventh transmembrane domain. RGR-opsin comes in different isoforms produced by alternative splicing.

X-linked retinitis pigmentosa GTPase regulator is a GTPase-binding protein that in humans is encoded by the RPGR gene. The gene is located on the X-chromosome and is commonly associated with X-linked retinitis pigmentosa (XLRP). In photoreceptor cells, RPGR is localized in the connecting cilium which connects the protein-synthesizing inner segment to the photosensitive outer segment and is involved in the modulation of cargo trafficked between the two segments.

Peripherin-2 is a protein, that in humans is encoded by the PRPH2 gene. Peripherin-2 is found in the rod and cone cells of the retina of the eye. Defects in this protein result in one form of retinitis pigmentosa, an incurable blindness.

PRP31 pre-mRNA processing factor 31 homolog , also known as PRPF31, is a protein which in humans is encoded by the PRPF31 gene.

Cone-rod homeobox protein is a protein that in humans is encoded by the CRX gene.
Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta is the beta subunit of the protein complex PDE6 that is encoded by the PDE6B gene. PDE6 is crucial in transmission and amplification of visual signal. The existence of this beta subunit is essential for normal PDE6 functioning. Mutations in this subunit are responsible for retinal degeneration such as retinitis pigmentosa or congenital stationary night blindness.

S-arrestin is a protein that in humans is encoded by the SAG gene.

Neural retina-specific leucine zipper protein is a protein that in humans is encoded by the NRL gene.

Tubby-related protein 1 is a protein that in humans is encoded by the TULP1 gene.

Rod outer segment membrane protein 1 is a protein that in humans is encoded by the ROM1 gene.

Oxygen-regulated protein 1 also known as retinitis pigmentosa 1 protein (RP1) is a protein that in humans is encoded by the RP1 gene.

Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit alpha is an enzyme that in humans is encoded by the PDE6A gene.

Fascin-2 is a protein that in humans is encoded by the FSCN2 gene.

Inosine-5'-monophosphate dehydrogenase 1, also known as IMP dehydrogenase 1, is an enzyme that in humans is encoded by the IMPDH1 gene.
Retinal gene therapy holds a promise in treating different forms of non-inherited and inherited blindness.




