Related Research Articles
A rare disease is a disease that affects a small percentage of the population. In some parts of the world, the term orphan disease describes a rare disease whose rarity results in little or no funding or research for treatments, without financial incentives from governments or other agencies. Orphan drugs are medications targeting orphan diseases.
In biochemistry, isomerases are a general class of enzymes that convert a molecule from one isomer to another. Isomerases facilitate intramolecular rearrangements in which bonds are broken and formed. The general form of such a reaction is as follows:
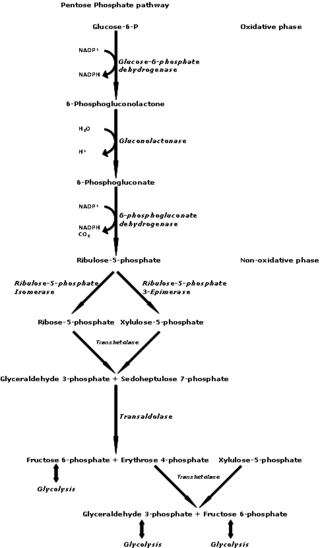
The pentose phosphate pathway is a metabolic pathway parallel to glycolysis. It generates NADPH and pentoses as well as ribose 5-phosphate, a precursor for the synthesis of nucleotides. While the pentose phosphate pathway does involve oxidation of glucose, its primary role is anabolic rather than catabolic. The pathway is especially important in red blood cells (erythrocytes). The reactions of the pathway were elucidated in the early 1950s by Bernard Horecker and co-workers.

Ribulose is a ketopentose — a monosaccharide containing five carbon atoms, and including a ketone functional group. It has chemical formula C5H10O5. Two enantiomers are possible, d-ribulose and l-ribulose. d-Ribulose is the diastereomer of d-xylulose.
Glucose-6-phosphate isomerase (GPI), alternatively known as phosphoglucose isomerase/phosphoglucoisomerase (PGI) or phosphohexose isomerase (PHI), is an enzyme that in humans is encoded by the GPI gene on chromosome 19. This gene encodes a member of the glucose phosphate isomerase protein family. The encoded protein has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions. In the cytoplasm, the gene product functions as a glycolytic enzyme that interconverts glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P). Extracellularly, the encoded protein functions as a neurotrophic factor that promotes survival of skeletal motor neurons and sensory neurons, and as a lymphokine that induces immunoglobulin secretion. The encoded protein is also referred to as autocrine motility factor (AMF) based on an additional function as a tumor-secreted cytokine and angiogenic factor. Defects in this gene are the cause of nonspherocytic hemolytic anemia, and a severe enzyme deficiency can be associated with hydrops fetalis, immediate neonatal death and neurological impairment. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2014]

Transketolase is an enzyme that, in humans, is encoded by the TKT gene. It participates in both the pentose phosphate pathway in all organisms and the Calvin cycle of photosynthesis. Transketolase catalyzes two important reactions, which operate in opposite directions in these two pathways. In the first reaction of the non-oxidative pentose phosphate pathway, the cofactor thiamine diphosphate accepts a 2-carbon fragment from a 5-carbon ketose (D-xylulose-5-P), then transfers this fragment to a 5-carbon aldose (D-ribose-5-P) to form a 7-carbon ketose (sedoheptulose-7-P). The abstraction of two carbons from D-xylulose-5-P yields the 3-carbon aldose glyceraldehyde-3-P. In the Calvin cycle, transketolase catalyzes the reverse reaction, the conversion of sedoheptulose-7-P and glyceraldehyde-3-P to pentoses, the aldose D-ribose-5-P and the ketose D-xylulose-5-P.

2-hydroxyglutaric aciduria is a rare neurometabolic disorder characterized by the significantly elevated levels of hydroxyglutaric acid in one's urine. It is either autosomal recessive or autosomal dominant.

Triosephosphate isomerase deficiency is a rare autosomal recessive metabolic disorder which was initially described in 1965.
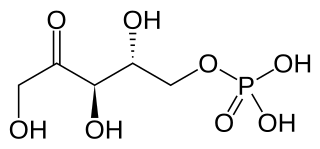
Ribulose 5-phosphate is one of the end-products of the pentose phosphate pathway. It is also an intermediate in the Calvin cycle.

Transaldolase is an enzyme of the non-oxidative phase of the pentose phosphate pathway. In humans, transaldolase is encoded by the TALDO1 gene.

Ribose 5-phosphate (R5P) is both a product and an intermediate of the pentose phosphate pathway. The last step of the oxidative reactions in the pentose phosphate pathway is the production of ribulose 5-phosphate. Depending on the body's state, ribulose 5-phosphate can reversibly isomerize to ribose 5-phosphate. Ribulose 5-phosphate can alternatively undergo a series of isomerizations as well as transaldolations and transketolations that result in the production of other pentose phosphates as well as fructose 6-phosphate and glyceraldehyde 3-phosphate.

Erythrose 4-phosphate is a phosphate of the simple sugar erythrose. It is an intermediate in the pentose phosphate pathway and the Calvin cycle.

6-Phosphogluconolactonase (EC 3.1.1.31, 6PGL, PGLS, systematic name 6-phospho-D-glucono-1,5-lactone lactonohydrolase) is a cytosolic enzyme found in all organisms that catalyzes the hydrolysis of 6-phosphogluconolactone to 6-phosphogluconic acid in the oxidative phase of the pentose phosphate pathway:

Ribose-5-phosphate isomerase (Rpi) encoded by the RPIA gene is an enzyme that catalyzes the conversion between ribose-5-phosphate (R5P) and ribulose-5-phosphate (Ru5P). It is a member of a larger class of isomerases which catalyze the interconversion of chemical isomers. It plays a vital role in biochemical metabolism in both the pentose phosphate pathway and the Calvin cycle. The systematic name of this enzyme class is D-ribose-5-phosphate aldose-ketose-isomerase.

Membrane protein MLC1 is a protein that in humans is encoded by the MLC1 gene.

D-2-hydroxyglutarate dehydrogenase, mitochondrial is an enzyme that in humans is encoded by the D2HGDH gene.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
Transaldolase deficiency is a disease characterised by abnormally low levels of the transaldolase enzyme. It is a metabolic enzyme involved in the pentose phosphate pathway. It is caused by mutation in the transaldolase gene (TALDO1). It was first described by Verhoeven et al. in 2001.
Megalencephalic leukoencephalopathy with subcortical cysts is a form of hereditary CNS demyelinating disease. It belongs to a group of disorders called leukodystrophies. It is characterized by early-onset enlargement of the head (macrocephaly) as well as delayed-onset neurological deterioration to include spasticity, epilepsy, and lack of muscular coordination. MLC does not appear to be a disease that is fatal at birth or early in life despite its symptoms, although the number of patients throughout history known to have the disease is fairly limited.

Transaldolase 1 is a protein that in humans is encoded by the TALDO1 gene.
References
- ↑ "OMIM Entry - # 608611 - Ribose 5-Phosphate Isomerase Deficiency". omim.org. Retrieved 16 March 2019.
- 1 2 Wamelink, M. M.; Grüning, N. M.; Jansen, E. E.; Bluemlein, K.; Lehrach, H.; Jakobs, C.; Ralser, M. (2010). "The difference between rare and exceptionally rare: molecular characterization of ribose 5-phosphate isomerase deficiency". J. Mol. Med. 88 (9): 931–39. doi:10.1007/s00109-010-0634-1. hdl:1871/34686. PMID 20499043. S2CID 10870492.
- ↑ Dalling, Robert (2017-02-10). "These twins are 'trapped' in their living room as work plans stall". WalesOnline. Retrieved 2021-07-31.
- 1 2 Huck JH, Verhoeven NM, Struys EA, Salomons GS, Jakobs C, van der Knaap MS (April 2004). "Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy". American Journal of Human Genetics. 74 (4): 745–51. doi:10.1086/383204. PMC 1181951 . PMID 14988808.
- ↑ Klusmann, A.; Fleischer, W.; Waldhaus, A.; Siebler, M.; Mayatepek, E. (December 2005). "Influence of D ‐arabitol and ribitol on neuronal network activity". Journal of Inherited Metabolic Disease. 28 (6): 1181–1183. doi:10.1007/s10545-005-0073-2. ISSN 0141-8955.
- ↑ Stone, V.; Kudo, K.Y.; August, P.M.; Marcelino, T.B.; Matté, C. (October 2014). "Polyols accumulated in ribose‐5‐phosphate isomerase deficiency increase mitochondrial superoxide production and improve antioxidant defenses in rats' prefrontal cortex". International Journal of Developmental Neuroscience. 37 (1): 21–25. doi:10.1016/j.ijdevneu.2014.06.009. ISSN 0736-5748.
- ↑ Wang, Ching‐Tzu; Chen, Yi‐Chun; Wang, Yi‐Yun; Huang, Ming‐Hao; Yen, Tzu‐Li; Li, Hsun; Liang, Cyong‐Jhih; Sang, Tzu‐Kang; Ciou, Shih‐Ci; Yuh, Chiou‐Hwa; Wang, Chao‐Yung; Brummel, Theodore J.; Wang, Horng‐Dar (February 2012). "Reduced neuronal expression of ribose‐5‐phosphate isomerase enhances tolerance to oxidative stress, extends lifespan, and attenuates polyglutamine toxicity in Drosophila". Aging Cell. 11 (1): 93–103. doi:10.1111/j.1474-9726.2011.00762.x. ISSN 1474-9718. PMC 3257417 . PMID 22040003.
- ↑ "Ribose 5-Phosphate Isomerase Deficiency disease: Malacards - Research Articles, Drugs, Genes, Clinical Trials". www.malacards.org. Retrieved 2018-03-05.
- 1 2 Huck, Jojanneke H. J.; Verhoeven, Nanda M.; Struys, Eduard A.; Salomons, Gajja S.; Jakobs, Cornelis; van der Knaap, Marjo S. (April 2004). "Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy". American Journal of Human Genetics. 74 (4): 745–751. doi:10.1086/383204. ISSN 0002-9297. PMC 1181951 . PMID 14988808.
- ↑ van der Knaap MS, Wevers RA, Struys EA, Verhoeven NM, Pouwels PJ, Engelke UF, Feikema W, Valk J, Jakobs C (December 1999). "Leukoencephalopathy associated with a disturbance in the metabolism of polyols". Annals of Neurology. 46 (6): 925–8. doi:10.1002/1531-8249(199912)46:6<925::aid-ana18>3.0.co;2-j. PMID 10589548. S2CID 43743595.
- ↑ Naik N, Shah A, Wamelink MC, van der Knaap MS, Hingwala D (September 2017). "Rare case of ribose 5 phosphate isomerase deficiency with slowly progressive leukoencephalopathy". Neurology. 89 (11): 1195–1196. doi:10.1212/WNL.0000000000004361. PMID 28801340. S2CID 9554949.
- ↑ Brooks SS, Anderson S, Bhise V, Botti C (October 2018). "Further Delineation of Ribose-5-phosphate Isomerase Deficiency: Report of a Third Case". Journal of Child Neurology. 33 (12): 784–787. doi:10.1177/0883073818789316. PMID 30088433. S2CID 51936427.
- ↑ Kaur, Parneet; Wamelink, Mirjam M.C.; Van Der Knaap, Marjo S.; Girisha, Katta Mohan; Shukla, Anju (2019-08-01). "Confirmation of a Rare Genetic Leukoencephalopathy due to a Novel Bi-allelic Variant in RPIA". European Journal of Medical Genetics. 62 (8): 103708. doi:10.1016/j.ejmg.2019.103708. ISSN 1769-7212. PMID 31247379. S2CID 195760193.