
Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
The Trans-Proteomic Pipeline (TPP) is an open-source data analysis software for proteomics developed at the Institute for Systems Biology (ISB) by the Ruedi Aebersold group under the Seattle Proteome Center. The TPP includes PeptideProphet, ProteinProphet, ASAPRatio, XPRESS and Libra.
In analytical chemistry, a tandem mass tag (TMT) is a chemical label that facilitates sample multiplexing in mass spectrometry (MS)-based quantification and identification of biological macromolecules such as proteins, peptides and nucleic acids. TMT belongs to a family of reagents referred to as isobaric mass tags which are a set of molecules with the same mass, but yield reporter ions of differing mass after fragmentation. The relative ratio of the measured reporter ions represents the relative abundance of the tagged molecule, although ion suppression has a detrimental effect on accuracy. Despite these complications, TMT-based proteomics has been shown to afford higher precision than label-free quantification. In addition to aiding in protein quantification, TMT tags can also increase the detection sensitivity of certain highly hydrophilic analytes, such as phosphopeptides, in RPLC-MS analyses.
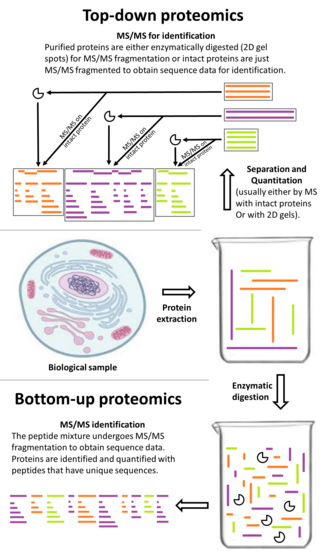
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.

Claudin-10 is a protein that in humans is encoded by the CLDN10 gene. It belongs to the group of claudins.

Claudin-20 is a protein that in humans is encoded by the CLDN20 gene. It belongs to the group of claudins.

Claudin-18 is a protein that in humans is encoded by the CLDN18 gene. It belongs to the group of claudins.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
In proteomics, GPM stands for "Global Proteome Machine". It is a web-based, open source user interface for analyzing and displaying protein identification data. It was originally designed by Rob Craig and Ron Beavis and first released in 2003. The interface creates a series of web browser page views of tandem mass spectrometry data that has been assigned to protein sequences. The underlying data documents are stored in BIOML format files.

Claudin-22 is a protein that in humans is encoded by the CLDN22 gene. It belongs to the group of claudins.
The Human Proteome Project (HPP) is a collaborative effort coordinated by the Human Proteome Organization. Its stated goal is to experimentally observe all of the proteins produced by the sequences translated from the human genome.
MassMatrix is a mass spectrometry data analysis software that uses a statistical model to achieve increased mass accuracy over other database search algorithms. This search engine is set apart from others dues to its ability to provide extremely efficient judgement between true and false positives for high mass accuracy data that has been obtained from present day mass spectrometer instruments. It is useful for identifying disulphide bonds in tandem mass spectrometry data. This search engine is set apart from others due to its ability to provide extremely efficient judgement between true and false positives for high mass accuracy data that has been obtained from present day mass spectrometer instruments.
David Fenyö is a Hungarian-Swedish-American computational biologist, physicist and businessman. He is currently professor in the Department of Biochemistry and Molecular Pharmacology at NYU Langone Medical Center. Fenyö's research focuses on the development of methods to identify, characterize and quantify proteins and in the integration of data from multiple modalities including mass spectrometry, sequencing and microscopy.
Skyline is an open source software for targeted proteomics and metabolomics data analysis. It runs on Microsoft Windows and supports the raw data formats from multiple mass spectrometric vendors. It contains a graphical user interface to display chromatographic data for individual peptide or small molecule analytes.

Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.
Debasis Dash is an Indian computational biologist and chief scientist at the Institute of Genomics and Integrative Biology (IGIB). Known for his research on proteomics and Big Data and Artificial Intelligence studies, his studies have been documented by way of a number of articles and ResearchGate, an online repository of scientific articles has listed 120 of them. The Department of Biotechnology of the Government of India awarded him the National Bioscience Award for Career Development, one of the highest Indian science awards, for his contributions to biosciences, in 2014. He was appointed as the director of Institute of Life Sciences, Bhubaneswar on 18 May 2023.
Young-Ki Paik (Korean: 백영기) is the director of the Yonsei Proteome Research Center in Seoul, Korea. In 2009, he was chosen President of the Human Proteome Organization (HUPO).
Ravi Sirdeshmukh is an Indian cancer biologist and proteomicist. He is a distinguished scientist and incumbent associate director of the Institute of Bioinformatics (IOB) in Bangalore, founding president of the Proteomics Society of India and senior research advisor at the Mazumdar Shaw Center for Translational Research (MSCTR) in Bangalore. He is also an elected member of the Council of Human Proteome Organization. He is most noted for his contributions in the Human Proteome Project where he served as the Group Leader for the countries like India, Singapore, Taiwan and Thailand. Sirdeshmukh is also an invited member of the Council of Asian Oceanean HUPO (AOHUPO).
Alex Kentsis is a Jewish-American scientist and physician at the Memorial Sloan Kettering Cancer Center and Weill Medical College of Cornell University, known for his contributions to understanding biological self-organization, protein folding, cell signaling, and cancer therapeutics.
