
Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
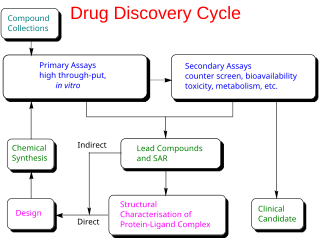
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
Quantitative structure–activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models relate a set of "predictor" variables (X) to the potency of the response variable (Y), while classification QSAR models relate the predictor variables to a categorical value of the response variable.
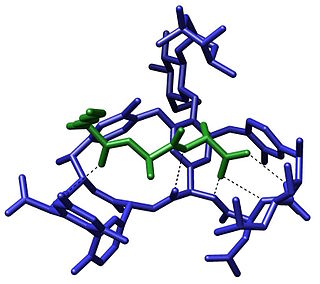
The term molecular recognition refers to the specific interaction between two or more molecules through noncovalent bonding such as hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, π-π interactions, halogen bonding, or resonant interaction effects. In addition to these direct interactions, solvents can play a dominant indirect role in driving molecular recognition in solution. The host and guest involved in molecular recognition exhibit molecular complementarity. Exceptions are molecular containers, including, e.g., nanotubes, in which portals essentially control selectivity. Selective partioning of molecules between two or more phases can also result in molecular recognition. In partitioning-based molecular recognition the kinetics and equilibrium conditions are governed by the presence of solutes in the two phases.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
SciTegic was a San Diego–based software company that developed and marketed informatics software to the pharmaceutical and biotechnology industries.
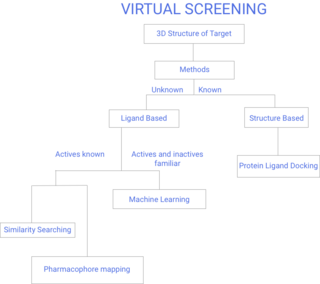
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
In the fields of computational chemistry and molecular modelling, scoring functions are mathematical functions used to approximately predict the binding affinity between two molecules after they have been docked. Most commonly one of the molecules is a small organic compound such as a drug and the second is the drug's biological target such as a protein receptor. Scoring functions have also been developed to predict the strength of intermolecular interactions between two proteins or between protein and DNA.
Inte:Ligand was founded in Maria Enzersdorf, Lower Austria (Niederösterreich) in 2003. They established the company headquarters on Mariahilferstrasse in Vienna, Austria that same year.
The program UCSF DOCK was created in the 1980s by Irwin "Tack" Kuntz's Group, and was the first docking program. DOCK uses geometric algorithms to predict the binding modes of small molecules. Brian K. Shoichet, David A. Case, and Robert C.Rizzo are codevelopers of DOCK.

FightAIDS@Home is a volunteer computing project operated by the Olson Laboratory at The Scripps Research Institute. It runs on internet-connected home computers, and since July 2013 also runs on Android smartphones and tablets. It aims to use biomedical software simulation techniques to search for ways to cure or prevent the spread of HIV/AIDS.
BindingDB is a public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of proteins considered to be candidate drug-targets with ligands that are small, drug-like molecules. As of March, 2011, BindingDB contains about 650,000 binding data, for 5,700 protein targets and 280,000 small molecules. BindingDB also includes a small collection of host–guest binding data of interest to chemists studying supramolecular systems.
Lead Finder is a computational chemistry tool designed for modelling protein-ligand interactions. It is used for conducting molecular docking studies and quantitatively assessing ligand binding and biological activity. It offers free access to users in commercial, academic, or other settings.
Druggability is a term used in drug discovery to describe a biological target that is known to or is predicted to bind with high affinity to a drug. Furthermore, by definition, the binding of the drug to a druggable target must alter the function of the target with a therapeutic benefit to the patient. The concept of druggability is most often restricted to small molecules but also has been extended to include biologic medical products such as therapeutic monoclonal antibodies.

SR-57227 is a potent and selective agonist at the 5HT3 receptor, with high selectivity over other serotonin receptor subtypes and good blood–brain barrier penetration.
Rommie E. Amaro is a professor and endowed chair of chemistry and biochemistry and the director of the National Biomedical Computation Resource at the University of California, San Diego. Her research focuses on development of computational methods in biophysics for applications to drug discovery.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.

David Weininger was an American cheminformatician and entrepreneur. He was most notable for inventing the chemical line notations for structures (SMILES), substructures (SMARTS) and reactions (SMIRKS). He also founded Daylight Chemical Information Systems, Inc.
Chimeric small molecule therapeutics are a class of drugs designed with multiple active domains to operate outside of the typical protein inhibition model. While most small molecule drugs inhibit target proteins by binding their active site, chimerics form protein-protein ternary structures to induce degradation or, less frequently, other protein modifications.