The enzyme encoded by SUCLG1 can exist in either a phosphorylated form or a dephosphorylated form. In the dephosphorylated structure, a phosphate ion works in coordination with a histidine residue in the active site and the two alpha helices, one contributed by each subunit of the alphabeta-dimer to stabilize the structure. One of the alpha helices contains amino acids, the modification of which result in conformational changes that accommodate either the bound phosphoryl group or the free phosphate ion.[7]
Function
This gene encodes the alpha subunit of the heterodimeric enzyme succinate coenzyme A ligase. This enzyme is targeted to the mitochondria and catalyzes the conversion of succinyl CoA and ADP or GDP to succinate and ATP or GTP. Mutations in this gene are the cause of the metabolic disorder fatal infantile lactic acidosis and mitochondrial DNA depletion.[8][9]
Clinical significance
Succinate-CoA ligase deficiency is responsible for encephalomyopathy with mitochondrial DNA depletion and mild methylmalonic aciduria. Mutations in SUCLG1 lead to complete absence of SUCLG1 protein and are responsible for a very severe disorder with antenatal manifestations. Furthermore, it is shown that in the absence of SUCLG1 protein, no SUCLA2 protein is found in fibroblasts by western blot analysis. This result is consistent with a degradation of SUCLA2 when its heterodimer partner, SUCLG1, is absent.[10] As mitochondrial DNA depletion in muscle is not a constant finding in SUCLG1 patients, diagnosis should not be based on it; additionally, it may be that alternative physiopathological mechanisms may be considered to explain the combined respiratory chain deficiency observed in these patients.[9]
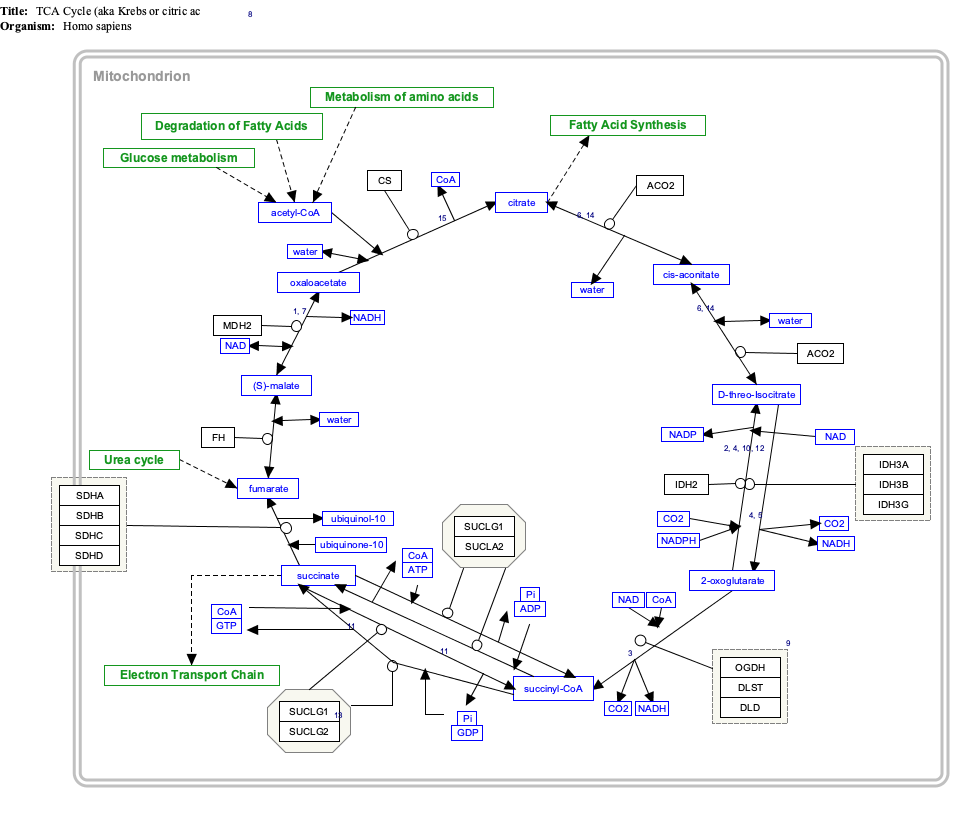
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
1 2 Valayannopoulos V, Haudry C, Serre V, Barth M, Boddaert N, Arnoux JB, Cormier-Daire V, Rio M, Rabier D, Vassault A, Munnich A, Bonnefont JP, de Lonlay P, Rötig A, Lebre AS (Jun 2010). "New SUCLG1 patients expanding the phenotypic spectrum of this rare cause of mild methylmalonic aciduria". Mitochondrion. 10 (4): 335–41. doi:10.1016/j.mito.2010.02.006. PMID20197121.
Maruyama K, Sugano S (Jan 1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID8125298.
Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S (Oct 1997). "Construction and characterization of a full length-enriched and a 5'-end-enriched cDNA library". Gene. 200 (1–2): 149–56. doi:10.1016/S0378-1119(97)00411-3. PMID9373149.
Fraser ME, James MN, Bridger WA, Wolodko WT (Jun 2000). "Phosphorylated and dephosphorylated structures of pig heart, GTP-specific succinyl-CoA synthetase". Journal of Molecular Biology. 299 (5): 1325–39. doi:10.1006/jmbi.2000.3807. PMID10873456.
Overview of all the structural information available in the PDB for UniProt: P53597 (Human Succinate--CoA ligase [ADP/GDP-forming] subunit alpha, mitochondrial) at the PDBe-KB.
Overview of all the structural information available in the PDB for UniProt: O19069 (Pig Succinate--CoA ligase [ADP/GDP-forming] subunit alpha, mitochondrial) at the PDBe-KB.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.