
Heme, or haem, is a precursor to hemoglobin, which is necessary to bind oxygen in the bloodstream. Heme is biosynthesized in both the bone marrow and the liver.

Cytochrome c peroxidase, or CCP, is a water-soluble heme-containing enzyme of the peroxidase family that takes reducing equivalents from cytochrome c and reduces hydrogen peroxide to water:
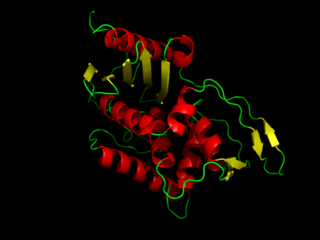
DD-transpeptidase is a bacterial enzyme that catalyzes the transfer of the R-L-αα-D-alanyl moiety of R-L-αα-D-alanyl-D-alanine carbonyl donors to the γ-OH of their active-site serine and from this to a final acceptor. It is involved in bacterial cell wall biosynthesis, namely, the transpeptidation that crosslinks the peptide side chains of peptidoglycan strands.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness. It was described by John Menkes in the 1950s.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Photosensitivity is an abnormal skin reaction to direct sunlight exposure. It is unrelated to a sunburn. These reactions are due to photosensitization, the accumulation of photosensitive compounds beneath the skin. In some cases, the photodynamic substances come from ingested plants or drugs, after being metabolized or not. In other cases, the photodynamic substances may be produced in the body itself due to inborn errors in pigment metabolism, especially those involving the heme synthesis. Photosensitivity reactions are usually seen in herbivorous or omnivorous animals, though such reactions are known among carnivores.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.

Ethylmalonic encephalopathy (EE) is a rare autosomal recessive inborn error of metabolism. Patients affected with EE are typically identified shortly after birth, with symptoms including diarrhea, petechiae and seizures. The genetic defect in EE is thought to involve an impairment in the degradation of sulfide intermediates in the body. Hydrogen sulfide then builds up to toxic levels. EE was initially described in 1994. Most cases of EE have been described in individuals of Mediterranean or Arabic origin.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

In enzymology, maleylacetoacetate isomerase is an enzyme that catalyzes the chemical reaction
In enzymology, an alanine—tRNA ligase is an enzyme that catalyzes the chemical reaction
Organic acidemia is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.

Fumarylacetoacetate hydrolase domain-containing protein 1, also known as FLJ36880 protein, is an enzyme that in humans is encoded by the FAHD1 gene on chromosome 16.

α-Ketoisocaproic acid (α-KIC), also known as 4-methyl-2-oxovaleric acid, and its conjugate base and carboxylate, α-ketoisocaproate, are metabolic intermediates in the metabolic pathway for L-leucine. Leucine is an essential amino acid, and its degradation is critical for many biological duties. α-KIC is produced in one of the first steps of the pathway by branched-chain amino acid aminotransferase by transferring the amine on L-leucine onto alpha ketoglutarate, and replacing that amine with a ketone. The degradation of L-leucine in the muscle to this compound allows for the production of the amino acids alanine and glutamate as well. In the liver, α-KIC can be converted to a vast number of compounds depending on the enzymes and cofactors present, including cholesterol, acetyl-CoA, isovaleryl-CoA, and other biological molecules. Isovaleryl-CoA is the main compound synthesized from ɑ-KIC. α-KIC is a key metabolite present in the urine of people with Maple syrup urine disease, along with other branched-chain amino acids. Derivatives of α-KIC have been studied in humans for their ability to improve physical performance during anaerobic exercise as a supplemental bridge between short-term and long-term exercise supplements. These studies show that α-KIC does not achieve this goal without other ergogenicsupplements present as well. α-KIC has also been observed to reduce skeletal muscle damage after eccentrically biased resistance exercises in people who do not usually perform those exercises.
Aminolevulinic acid dehydratase deficiency porphyria is an extremely rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). Lead poisoning can also disrupt ALAD and result in elevated ALA causing the same symptoms. Heme is a component of hemoglobin which carries oxygen in red blood cells.

Phenolic acids or phenolcarboxylic acids are types of aromatic acid compounds. Included in that class are substances containing a phenolic ring and an organic carboxylic acid function. Two important naturally occurring types of phenolic acids are hydroxybenzoic acids and hydroxycinnamic acids, which are derived from non-phenolic molecules of benzoic and cinnamic acid, respectively.
4-Hydroxyphenylpyruvate dioxygenase (HPPD) inhibitors are a class of herbicides that prevent growth in plants by blocking 4-Hydroxyphenylpyruvate dioxygenase, an enzyme in plants that breaks down the amino acid tyrosine into molecules that are then used by plants to create other molecules that plants need. This process of breakdown, or catabolism, and making new molecules from the results, or biosynthesis, is something all living things do. HPPD inhibitors were first brought to market in 1980, although their mechanism of action was not understood until the late 1990s. They were originally used primarily in Japan in rice production, but since the late 1990s have been used in Europe and North America for corn, soybeans, and cereals, and since the 2000s have become more important as weeds have become resistant to glyphosate and other herbicides. Genetically modified crops are under development that include resistance to HPPD inhibitors. There is a pharmaceutical drug on the market, nitisinone, that was originally under development as an herbicide as a member of this class, and is used to treat an orphan disease, type I tyrosinemia.
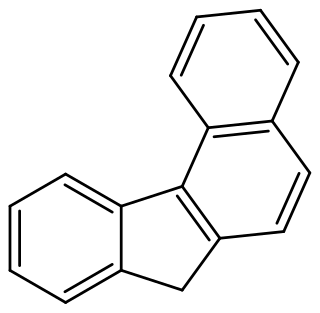
Benzo[c]fluorene is a polycyclic aromatic hydrocarbon (PAH) with mutagenic activity. It is a component of coal tar, cigarette smoke and smog and thought to be a major contributor to its carcinogenic properties. The mutagenicity of benzo[c]fluorene is mainly attributed to formation of metabolites that are reactive and capable of forming DNA adducts. According to the KEGG it is a group 3 carcinogen. Other names for benzo[c]fluorene are 7H-benzo[c]fluorene, 3,4-benzofluorene, and NSC 89264.

Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves. The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. If not detected via newborn screening and management not begun before symptoms appear, clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe. If diagnosed through newborn screening prior to clinical manifestation, and well managed with diet and medication, normal growth and development is possible.
