Protein primary structure is the linear sequence of amino acids in a peptide or protein. By convention, the primary structure of a protein is reported starting from the amino-terminal (N) end to the carboxyl-terminal (C) end. Protein biosynthesis is most commonly performed by ribosomes in cells. Peptides can also be synthesized in the laboratory. Protein primary structures can be directly sequenced, or inferred from DNA sequences.

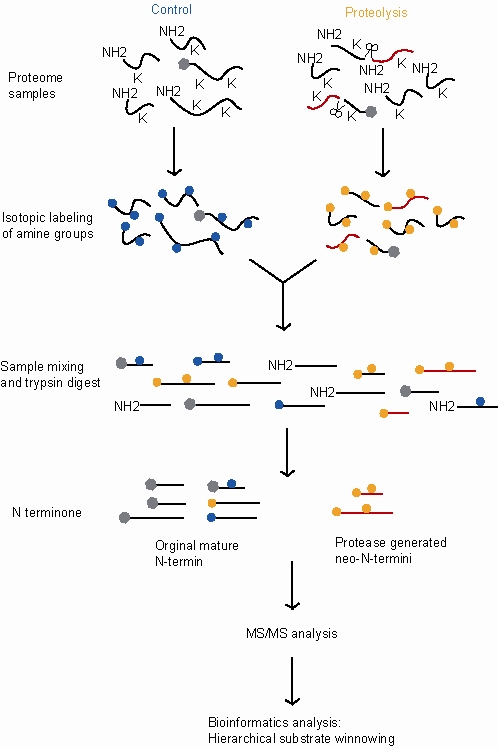
Proteolysis is the breakdown of proteins into smaller polypeptides or amino acids. Uncatalysed, the hydrolysis of peptide bonds is extremely slow, taking hundreds of years. Proteolysis is typically catalysed by cellular enzymes called proteases, but may also occur by intra-molecular digestion.

Trypsin is a serine protease from the PA clan superfamily, found in the digestive system of many vertebrates, where it hydrolyzes proteins. Trypsin is formed in the small intestine when its proenzyme form, the trypsinogen produced by the pancreas, is activated. Trypsin cuts peptide chains mainly at the carboxyl side of the amino acids lysine or arginine. It is used for numerous biotechnological processes. The process is commonly referred to as trypsin proteolysis or trypsinization, and proteins that have been digested/treated with trypsin are said to have been trypsinized. Trypsin was discovered in 1876 by Wilhelm Kühne and was named from the Ancient Greek word for rubbing since it was first isolated by rubbing the pancreas with glycerin.

A protease is an enzyme that catalyzes proteolysis, the breakdown of proteins into smaller polypeptides or single amino acids. They do this by cleaving the peptide bonds within proteins by hydrolysis, a reaction where water breaks bonds. Proteases are involved in many biological functions, including digestion of ingested proteins, protein catabolism, and cell signaling.
The C-terminus is the end of an amino acid chain, terminated by a free carboxyl group (-COOH). When the protein is translated from messenger RNA, it is created from N-terminus to C-terminus. The convention for writing peptide sequences is to put the C-terminal end on the right and write the sequence from N- to C-terminus.
The N-terminus (also known as the amino-terminus, NH2-terminus, N-terminal end or amine-terminus) is the start of a protein or polypeptide referring to the free amine group (-NH2) located at the end of a polypeptide. Within a peptide, the amine group is bonded to another carboxylic group in a protein to make it a chain, but since the end amino acid of a protein is only connected at the carboxy- end, the remaining free amine group is called the N-terminus. By convention, peptide sequences are written N-terminus to C-terminus, left to right (in LTR writing systems). This correlates the translation direction to the text direction (because when a protein is translated from messenger RNA, it is created from N-terminus to C-terminus - amino acids are added to the carboxyl end).

Protein sequencing is the practical process of determining the amino acid sequence of all or part of a protein or peptide. This may serve to identify the protein or characterize its post-translational modifications. Typically, partial sequencing of a protein provides sufficient information to identify it with reference to databases of protein sequences derived from the conceptual translation of genes.

Stable Isotope Labeling by/with Amino acids in Cell culture (SILAC) is a technique based on mass spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling. It is a popular method for quantitative proteomics.
PEAKS is a proteomics software program for tandem mass spectrometry designed for peptide sequencing, protein identification and quantification.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE) or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
The in-gel digestion step is a part of the sample preparation for the mass spectrometric identification of proteins in course of proteomic analysis. The method was introduced in 1992 by Rosenfeld. Innumerable modifications and improvements in the basic elements of the procedure remain.
Label-free quantification is a method in mass spectrometry that aims to determine the relative amount of proteins in two or more biological samples. Unlike other methods for protein quantification, label-free quantification does not use a stable isotope containing compound to chemically bind to and thus label the protein.
An Isotope-coded affinity tag (ICAT) is an in-vitro isotopic labeling method used for quantitative proteomics by mass spectrometry that uses chemical labeling reagents. These chemical probes consist of three elements: a reactive group for labeling an amino acid side chain, an isotopically coded linker, and a tag for the affinity isolation of labeled proteins/peptides. The samples are combined and then separated through chromatography, then sent through a mass spectrometer to determine the mass-to-charge ratio between the proteins. Only cysteine containing peptides can be analysed. Since only cysteine containing peptides are analysed, often the post translational modification is lost.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with various chemical groups that are identical masses (isobaric), but vary in terms of distribution of heavy isotopes around their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy CID (HCD) during tandem mass spectrometry yielding reporter ions of different masses. The most common isobaric tags are amine-reactive tags. However, tags that react with cysteine residues and carbonyl groups have also been described. These amine-reactive groups go through N-hydroxysuccinimide (NHS) reactions, which are based around three types of functional groups. Isobaric labeling methods include tandem mass tags (TMT), isobaric tags for relative and absolute quantification (iTRAQ), mass differential tags for absolute and relative quantification, and dimethyl labeling. TMTs and iTRAQ methods are most common and developed of these methods. Tandem mass tags have a mass reporter region, a cleavable linker region, a mass normalization region, and a protein reactive group and have the same total mass.
TopFIND is the Termini oriented protein Function Inferred Database (TopFIND) is an integrated knowledgebase focused on protein termini, their formation by proteases and functional implications. It contains information about the processing and the processing state of proteins and functional implications thereof derived from research literature, contributions by the scientific community and biological databases.

Degradomics is a sub-discipline of biology encompassing all the genomic and proteomic approaches devoted to the study of proteases, their inhibitors, and their substrates on a system-wide scale. This includes the analysis of the protease and protease-substrate repertoires, also called "protease degradomes". The scope of these degradomes can range from cell, tissue, and organism-wide scales.
Stable isotope standards and capture by anti-peptide antibodies (SISCAPA) is a mass spectrometry method for measuring the amount of a protein in a biological sample.

Translatomics is the study of all open reading frames (ORFs) that are being actively translated in a cell or organism. This collection of ORFs is called the translatome. Characterizing a cell's translatome can give insight into the array of biological pathways that are active in the cell. According to the central dogma of molecular biology, the DNA in a cell is transcribed to produce RNA, which is then translated to produce a protein. Thousands of proteins are encoded in an organism's genome, and the proteins present in a cell cooperatively carry out many functions to support the life of the cell. Under various conditions, such as during stress or specific timepoints in development, the cell may require different biological pathways to be active, and therefore require a different collection of proteins. Depending on intrinsic and environmental conditions, the collection of proteins being made at one time varies. Translatomic techniques can be used to take a "snapshot" of this collection of actively translating ORFs, which can give information about which biological pathways the cell is activating under the present conditions.