Heat shock proteins (HSPs) are a family of proteins produced by cells in response to exposure to stressful conditions. They were first described in relation to heat shock, but are now known to also be expressed during other stresses including exposure to cold, UV light and during wound healing or tissue remodeling. Many members of this group perform chaperone functions by stabilizing new proteins to ensure correct folding or by helping to refold proteins that were damaged by the cell stress. This increase in expression is transcriptionally regulated. The dramatic upregulation of the heat shock proteins is a key part of the heat shock response and is induced primarily by heat shock factor (HSF). HSPs are found in virtually all living organisms, from bacteria to humans.
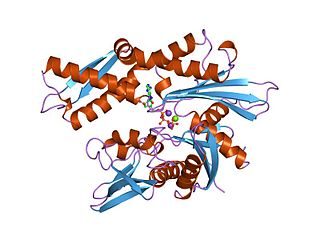
The 70 kilodalton heat shock proteins are a family of conserved ubiquitously expressed heat shock proteins. Proteins with similar structure exist in virtually all living organisms. Intracellularly localized Hsp70s are an important part of the cell's machinery for protein folding, performing chaperoning functions, and helping to protect cells from the adverse effects of physiological stresses. Additionally, membrane-bound Hsp70s have been identified as a potential target for cancer therapies and their extracellularly localized counterparts have been identified as having both membrane-bound and membrane-free structures.
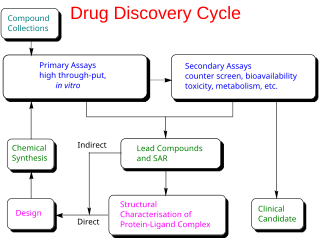
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.

Fatty-acid amide hydrolase 1 (FAAH) is a member of the serine hydrolase family of enzymes. It was first shown to break down anandamide (AEA), an N-acylethanolamine (NAE) in 1993. In humans, it is encoded by the gene FAAH.
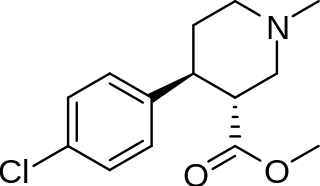
(+)-CPCA is a stimulant drug similar in structure to pethidine and to RTI-31, but nocaine is lacking the two-carbon bridge of RTI-31's tropane skeleton. This compound was first developed as a substitute agent for cocaine.

Heat shock protein HSP 90-alpha is a protein that in humans is encoded by the HSP90AA1 gene.
Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD) is a method used for finding lead compounds as part of the drug discovery process. Fragments are small organic molecules which are small in size and low in molecular weight. It is based on identifying small chemical fragments, which may bind only weakly to the biological target, and then growing them or combining them to produce a lead with a higher affinity. FBLD can be compared with high-throughput screening (HTS). In HTS, libraries with up to millions of compounds, with molecular weights of around 500 Da, are screened, and nanomolar binding affinities are sought. In contrast, in the early phase of FBLD, libraries with a few thousand compounds with molecular weights of around 200 Da may be screened, and millimolar affinities can be considered useful. FBLD is a technique being used in research for discovering novel potent inhibitors. This methodology could help to design multitarget drugs for multiple diseases. The multitarget inhibitor approach is based on designing an inhibitor for the multiple targets. This type of drug design opens up new polypharmacological avenues for discovering innovative and effective therapies. Neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s, among others, also show rather complex etiopathologies. Multitarget inhibitors are more appropriate for addressing the complexity of AD and may provide new drugs for controlling the multifactorial nature of AD, stopping its progression.

An Hsp90 inhibitor is a substance that inhibits that activity of the Hsp90 heat shock protein. Since Hsp90 stabilizes a variety of proteins required for survival of cancer cells, these substances may have therapeutic benefit in the treatment of various types of malignancies. Furthermore, a number of Hsp90 inhibitors are currently undergoing clinical trials for a variety of cancers. Hsp90 inhibitors include the natural products geldanamycin, Retaspimycin hydrochloride and radicicol as well as semisynthetic derivatives 17-N-Allylamino-17-demethoxygeldanamycin (17AAG).
Druggability is a term used in drug discovery to describe a biological target that is known to or is predicted to bind with high affinity to a drug. Furthermore, by definition, the binding of the drug to a druggable target must alter the function of the target with a therapeutic benefit to the patient. The concept of druggability is most often restricted to small molecules but also has been extended to include biologic medical products such as therapeutic monoclonal antibodies.
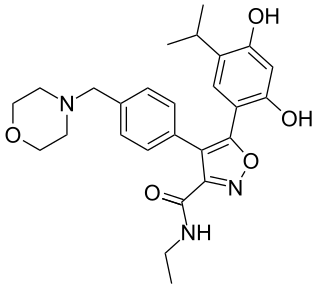
Luminespib is an experimental drug candidate for the treatment of cancer. It was discovered through a collaboration between The Institute of Cancer Research and the pharmaceutical company Vernalis and licensed to Novartis. From 2011 to 2014 it was in Phase II clinical trials. Chemically it is a resorcinylic isoxazole amide

Paul Workman is a British scientist noted for his work on the discovery and development of pharmaceutical agents in the field of oncology. He is President and CEO of The Institute of Cancer Research In London.
JZP150 (formerly PF-04457845) is an inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50 of 7.2nM, and both analgesic and antiinflammatory effects in animal studies comparable to naproxen.

mTOR inhibitors are a class of drugs used to treat several human diseases, including cancer, autoimmune diseases, and neurodegeneration. They function by inhibiting the mammalian target of rapamycin (mTOR), which is a serine/threonine-specific protein kinase that belongs to the family of phosphatidylinositol-3 kinase (PI3K) related kinases (PIKKs). mTOR regulates cellular metabolism, growth, and proliferation by forming and signaling through two protein complexes, mTORC1 and mTORC2. The most established mTOR inhibitors are so-called rapalogs, which have shown tumor responses in clinical trials against various tumor types.
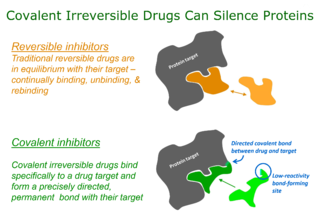
Targeted covalent inhibitors (TCIs) or Targeted covalent drugs are rationally designed inhibitors that bind and then bond to their target proteins. These inhibitors possess a bond-forming functional group of low chemical reactivity that, following binding to the target protein, is positioned to react rapidly with a proximate nucleophilic residue at the target site to form a bond.

MK-4409 is an experimental drug which acts as a potent and selective inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50 of 11 nM, and both analgesic and antiinflammatory effects in animal studies. It was studied for the treatment of neuropathic pain and progressed to early stage human clinical trials by 2009.

JNJ-42165279 is a drug developed by Janssen Pharmaceutica which acts as a potent and selective inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50 of 70 nM. It is described as a covalently binding but slowly reversible selective inhibitor of FAAH. JNJ-42165279 is being developed for the treatment of anxiety disorders and major depressive disorder. Clinical development has progressed as far as Phase II human trials with two studies in patients with mood disorders registered in ClinicalTrials.gov.
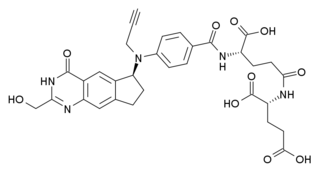
ONX-0801 is an experimental drug that has been developed to target ovarian cancer. It is a folate receptor alpha mediated thymidylate synthase inhibitor.

XL-388 is a drug which acts as a potent and selective inhibitor of both subtypes of the mechanistic target of rapamycin (mTOR), mTORC1 and mTORC2. It is being researched for the treatment of various forms of cancer, and has also been used to demonstrate a potential application for mTOR inhibitors in the treatment of neuropathic pain.

Torin-1 is a drug which was one of the first non-rapalog derived inhibitors of the mechanistic target of rapamycin (mTOR) subtypes mTORC1 and mTORC2. In animal studies it has anti-inflammatory, anti-cancer, and anti-aging properties, and shows activity against neuropathic pain.
