The enzyme Uridine monophosphate synthase catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. In humans, the gene that codes for this enzyme is located on the long arm of chromosome 3 (3q13).
A salvage pathway is a pathway in which a biological product is produced from intermediates in the degradative pathway of its own or a similar substance. The term often refers to nucleotide salvage in particular, in which nucleotides are synthesized from intermediates in their degradative pathway.
A nucleoside triphosphate is a nucleoside containing a nitrogenous base bound to a 5-carbon sugar, with three phosphate groups bound to the sugar. They are the molecular precursors of both DNA and RNA, which are chains of nucleotides made through the processes of DNA replication and transcription. Nucleoside triphosphates also serve as a source of energy for cellular reactions and are involved in signalling pathways.

Lesch–Nyhan syndrome (LNS) is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This deficiency occurs due to mutations in the HPRT1 gene located on the X chromosome. LNS affects about 1 in 380,000 live births. The disorder was first recognized and clinically characterized by American medical student Michael Lesch and his mentor, pediatrician William Nyhan, at Johns Hopkins.

Adenosine deaminase is an enzyme involved in purine metabolism. It is needed for the breakdown of adenosine from food and for the turnover of nucleic acids in tissues.

HAT Medium is a selection medium for mammalian cell culture, which relies on the combination of aminopterin, a drug that acts as a powerful folate metabolism inhibitor by inhibiting dihydrofolate reductase, with hypoxanthine and thymidine which are intermediates in DNA synthesis. The trick is that aminopterin blocks DNA de novo synthesis, which is absolutely required for cell division to proceed, but hypoxanthine and thymidine provide cells with the raw material to evade the blockage, provided that they have the right enzymes, which means having functioning copies of the genes that encode them.
Adenine phosphoribosyltransferase (APRTase) is an enzyme encoded by the APRT gene, found in humans on chromosome 16. It is part of the Type I PRTase family and is involved in the nucleotide salvage pathway, which provides an alternative to nucleotide biosynthesis de novo in humans and most other animals. In parasitic protozoa such as giardia, APRTase provides the sole mechanism by which AMP can be produced. APRTase deficiency contributes to the formation of kidney stones (urolithiasis) and to potential kidney failure.

Adenine phosphoribosyltransferase deficiency is a rare autosomal recessive metabolic disorder caused by mutations of the APRT gene. Adenine phosphoribosyltransferase (APRT) catalyzes the creation of pyrophosphate and adenosine monophosphate from 5-phosphoribosyl-1-pyrophosphate and adenine. Adenine phosphoribosyltransferase is a purine salvage enzyme. Genetic mutations of adenine phosphoribosyltransferase make large amounts of 2,8-Dihydroxyadenine causing urolithiasis and renal failure.
Purine nucleoside phosphorylase, PNP, PNPase or inosine phosphorylase is an enzyme that in humans is encoded by the NP gene. It catalyzes the chemical reaction
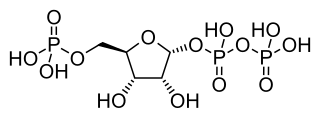
Phosphoribosyl pyrophosphate (PRPP) is a pentose phosphate. It is a biochemical intermediate in the formation of purine nucleotides via inosine-5-monophosphate, as well as in pyrimidine nucleotide formation. Hence it is a building block for DNA and RNA. The vitamins thiamine and cobalamin, and the amino acid tryptophan also contain fragments derived from PRPP. It is formed from ribose 5-phosphate (R5P) by the enzyme ribose-phosphate diphosphokinase:

Nucleic acid metabolism is a collective term that refers to the variety of chemical reactions by which nucleic acids are either synthesized or degraded. Nucleic acids are polymers made up of a variety of monomers called nucleotides. Nucleotide synthesis is an anabolic mechanism generally involving the chemical reaction of phosphate, pentose sugar, and a nitrogenous base. Degradation of nucleic acids is a catabolic reaction and the resulting parts of the nucleotides or nucleobases can be salvaged to recreate new nucleotides. Both synthesis and degradation reactions require multiple enzymes to facilitate the event. Defects or deficiencies in these enzymes can lead to a variety of diseases.
J(arvis) Edwin Seegmiller, or Jay Seegmiller, was an American physician and biochemical geneticist best known for his role in discovering the biochemical basis of the Lesch–Nyhan syndrome. He was a rheumatologist and a pioneer in research on arthritic diseases and on aging.

Ribose 5-phosphate (R5P) is both a product and an intermediate of the pentose phosphate pathway. The last step of the oxidative reactions in the pentose phosphate pathway is the production of ribulose 5-phosphate. Depending on the body's state, ribulose 5-phosphate can reversibly isomerize to ribose 5-phosphate. Ribulose 5-phosphate can alternatively undergo a series of isomerizations as well as transaldolations and transketolations that result in the production of other pentose phosphates as well as fructose 6-phosphate and glyceraldehyde 3-phosphate.

Purine nucleoside phosphorylase deficiency is a rare autosomal recessive metabolic disorder which results in immunodeficiency.
Purine metabolism refers to the metabolic pathways to synthesize and break down purines that are present in many organisms.

Amidophosphoribosyltransferase (ATase), also known as glutamine phosphoribosylpyrophosphate amidotransferase (GPAT), is an enzyme responsible for catalyzing the conversion of 5-phosphoribosyl-1-pyrophosphate (PRPP) into 5-phosphoribosyl-1-amine (PRA), using the amine group from a glutamine side-chain. This is the committing step in de novo purine synthesis. In humans it is encoded by the PPAT gene. ATase is a member of the purine/pyrimidine phosphoribosyltransferase family.
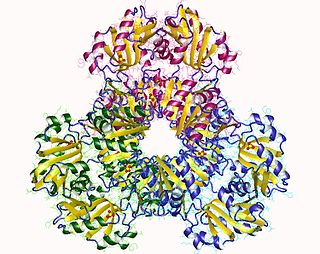
Ribose-phosphate diphosphokinase is an enzyme that converts ribose 5-phosphate into phosphoribosyl pyrophosphate (PRPP). It is classified under EC 2.7.6.1.
Somatic fusion, also called protoplast fusion, is a type of genetic modification in plants by which two distinct species of plants are fused together to form a new hybrid plant with the characteristics of both, a somatic hybrid. Hybrids have been produced either between different varieties of the same species or between two different species.
Nobuyo N. Maeda is a Japanese geneticist and medical researcher, who works on complex human diseases including atherosclerosis, diabetes and high blood pressure, and is particularly known for creating the first mouse model for atherosclerosis. Maeda has worked in the United States since 1978; as of 2017, she is the Robert H. Wagner Distinguished Professor at the University of North Carolina at Chapel Hill.

Arts syndrome is a rare metabolic disorder that causes serious neurological problems in males due to a malfunction of the PRPP synthetase 1 enzyme. Arts Syndrome is part of a spectrum of PRPS-1 related disorders with reduced activity of the enzyme that includes Charcot–Marie–Tooth disease and X-linked non-syndromic sensorineural deafness.




