Method of separating enantiomers in a racemic mixture by reaction rate
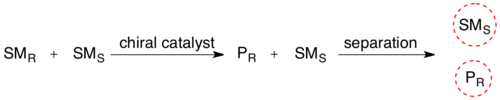
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer.[1] As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes.
Kinetic resolution can be used for the preparation of chiral molecules in organic synthesis. Kinetic resolution reactions utilizing purely synthetic reagents and catalysts are much less common than the use of enzymatic kinetic resolution in application towards organic synthesis, although a number of useful synthetic techniques have been developed in the past 30 years.[2]
History
The first reported kinetic resolution was achieved by Louis Pasteur. After reacting aqueous racemic ammonium tartrate with a mold from Penicillium glaucum, he reisolated the remaining tartrate and found it was levorotatory.[3] The chiral microorganisms present in the mold catalyzed the metabolization of (R,R)-tartrate selectively, leaving an excess of (S,S)-tartrate.
Kinetic resolution by synthetic means was first reported by Marckwald and McKenzie in 1899 in the esterification of racemicmandelic acid with optically active (−)-menthol. With an excess of the racemic acid present, they observed the formation of the ester derived from (+)-mandelic acid to be quicker than the formation of the ester from (−)-mandelic acid. The unreacted acid was observed to have a slight excess of (−)-mandelic acid, and the ester was later shown to yield (+)-mandelic acid upon saponification. The importance of this observation was that, in theory, if a half equivalent of (−)-menthol had been used, a highly enantioenriched sample of (−)-mandelic acid could have been prepared. This observation led to the successful kinetic resolution of other chiral acids, the beginning of the use of kinetic resolution in organic chemistry.[4][5]
Theory
Kinetic resolution is a possible method for irreversibly differentiating a pair of enantiomers due to (potentially) different activation energies. While both enantiomers are at the same Gibbs free energy level by definition, and the products of the reaction with both enantiomers are also at equal levels, the , or transition state energy, can differ. In the image below, the R enantiomer has a lower and would thus react faster than the S enantiomer.
The ideal kinetic resolution is that in which only one enantiomer reacts, i.e. kR>>kS. The selectivity (s) of a kinetic resolution is related to the rate constants of the reaction of the R and S enantiomers, kR and kS respectively, by s=kR/kS, for kR>kS. This selectivity can also be referred to as the relative rates of reaction. This can be written in terms of the free energy difference between the high- and low-energy transitions states, .[6]
The selectivity can also be expressed in terms of ee of the recovered starting material and conversion (c), if first-order kinetics (in substrate) are assumed. If it is assumed that the S enantiomer of the starting material racemate will be recovered in excess, it is possible to express the concentrations (mole fractions) of the S and R enantiomers as
where ee is the ee of the starting material. Note that for c=0, which signifies the beginning of the reaction, , where these signify the initial concentrations of the enantiomers. Then, for stoichiometric chiral resolving agent B*,
Note that, if the resolving agent is stoichiometric and achiral, with a chiral catalyst, the [B*] term does not appear. Regardless, with a similar expression for R, we can express s as
If we wish to express this in terms of the enantiomeric excess of the product, ee", we must make use of the fact that, for products R' and S' from R and S, respectively
From here, we see that
which gives us
which, when we plug into our expression for s derived above, yield
The conversion (c) and selectivity factor (s) can be expressed in terms of starting material and product enantiomeric excesses (ee and ee'', respectively) only:
Additionally, the expressions for c and ee can be parametrized to give explicit expressions for C and ee in terms of t. First, solving explicitly for [S] and [R] as functions of t yields
which, plugged into expressions for ee and c, gives
Without loss of generality, we can allow kS=1, which gives kR=s, simplifying the above expressions. Similarly, an expression for ee″ as a function of t can be derived
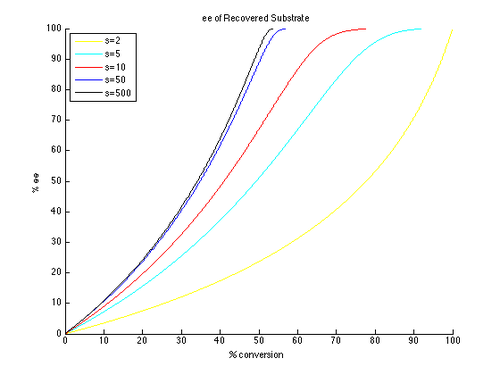
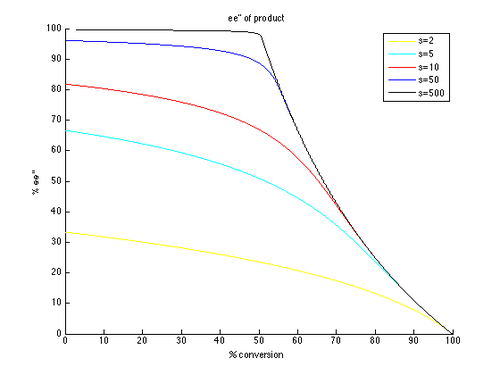
Thus, plots of ee and ee″ vs. c can be generated with t as the parameter and different values of s generating different curves, as shown below.
As can be seen, high enantiomeric excesses are much more readily attainable for the unreacted starting material. There is however a tradeoff between ee and conversion, with higher ee (of the recovered substrate) obtained at higher conversion, and therefore lower isolated yield. For example, with a selectivity factor of just 10, 99% ee is possible with approximately 70% conversion, resulting in a yield of about 30%. In contrast, in order to get good ee's and yield of the product, very high selectivity factors are necessary. For example, with a selectivity factor of 10, ee″ above approximately 80% is unattainable, and significantly lower ee″ values are obtained for more realistic conversions. A selectivity in excess of 50 is required for highly enantioenriched product, in reasonable yield.
This is a simplified version of the true kinetics of kinetic resolution. The assumption that the reaction is first order in substrate is limiting, and it is possible that the dependence on substrate may depend on conversion, resulting in a much more complicated picture. As a result, a common approach is to measure and report only yields and ee's, as the formula for krel only applies to an idealized kinetic resolution. It is simple to consider an initial substrate-catalyst complex forming, which could negate the first-order kinetics. However, the general conclusions drawn are still helpful to understand the effect of selectivity and conversion on ee.
Practicality
With the advent of asymmetric catalysis, it is necessary to consider the practicality of utilizing kinetic resolution for the preparation of enantiopure products. Even for a product which can be attained through an asymmetric catalytic or auxiliary-based route, the racemate may be significantly less expensive than the enantiopure material, resulting in heightened cost-effectiveness even with the inherent "loss" of 50% of the material. The following have been proposed as necessary conditions for a practical kinetic resolution:[6]
resolution proceeds selectively at low catalyst loadings
separation of starting material and product is easy
To date, a number of catalysts for kinetic resolution have been developed that satisfy most, if not all of the above criteria, making them highly practical for use in organic synthesis. The following sections will discuss a number of key examples.
Reactions utilizing synthetic reagents
Acylation reactions
Fu's planar chiral DMAP (-)-catalyst for the kinetic resolution of secondary alcohols
Gregory Fu and colleagues have developed a methodology utilizing a chiral DMAP analogue to achieve excellent kinetic resolution of secondary alcohols.[7] Initial studies utilizing ether as a solvent, low catalyst loadings (2mol%), acetic anhydride as the acylating agent, and triethylamine at room temperature gave selectivities ranging from 14-52, corresponding to ee's of the recovered alcohol product as high as 99.2%.[8] However, solvent screening proved that the use of tert-amyl alcohol increased both the reactivity and selectivity.[9]
With the benchmark substrate 1-phenylethanol, this corresponded to 99% ee of the unreacted alcohol at 55% conversion when run at 0°C. This system proved to be adept at resolution of a number of arylalkylcarbinols, with selectivities as high as 95 and low catalyst loadings of 1%, as shown below utilizing the (-)-enantiomer of the catalyst. This resulted in highly enantioenriched alcohols at very low conversions, giving excellent yields as well. In addition, the high selectivities result in highly enantioenriched acylated products, with a 90% ee sample of acylated alcohol for o-tolylmethylcarbinol, with s=71.
In addition, Fu reported the first highly selective acylation of racemic diols (as well as desymmetrization of meso diols). With low catalyst loading of 1%, enantioenriched diol was recovered in 98% ee and 43% yield, with the diacetate in 39% yield and 99% ee. The remainder of the material was recovered as a mixture of monoacetate.
The planar-chiral DMAP catalyst was also shown to be effective at kinetically resolving propargylic alcohols.[10] In this case, though, selectivities were found to be highest without any base present. When run with 1mol% of the catalyst at 0°C, selectivities as high as 20 could be attained. The limitations of this method include the requirement of an unsaturated functionality, such as carbonyl or alkenes, at the remote alkynyl position. Alcohols resolved using the (+)-enantiomer of the DMAP catalyst are shown below.
Fu also showed his chiral DMAP catalyst's ability to resolve allylic alcohols.[11] Effective selectivity was dependent upon the presence of either a geminal or cis substituent to the alcohol-bearing group, with a notable exception of a trans-phenyl alcohol which exhibited the highest selectivity. Using 1-2.5mol% of the (+)-enantiomer of the DMAP catalyst, the alcohols shown below were resolved in the presence of triethylamine.
Fu's (-)-PPY* catalyst (left) and novel acylating agent (right)
While Fu's DMAP analogue catalyst worked exceptionally well to kinetically resolve racemic alcohols, it was not successful in use for the kinetic resolution of amines. A similar catalyst, PPY*, was developed that, in use with a novel acylating agent, allowed for the successful kinetic resolution acylation of amines. With 10mol% (-)-PPY* in chloroform at –50°C, good to very good selectivities were observed in the acylation of amines, shown below.[12] A similar protocol was developed for the kinetic resolution of indolines.[13]
Epoxidations and dihydroxylations
The Sharpless epoxidation, developed by K. Barry Sharpless in 1980,[14] has been utilized for the kinetic resolution of a racemic mixture of allylic alcohols.[15][16] While extremely effective at resolving a number of allylic alcohols, this method has a number of drawbacks. Reaction times can run as long as 6 days, and the catalyst is not recyclable. However, the Sharpless asymmetric epoxidation kinetic resolution remains one of the most effective synthetic kinetic resolutions to date. A number of different tartrates can be used for the catalyst; a representative scheme is shown below utilizing diisopropyl tartrate. This method has seen general use on a number of secondary allylic alcohols.[17]
Sharpless asymmetric dihydroxylation has also seen use as a method for kinetic resolution.[18][19] This method is not widely used, however, since the same resolution can be accomplished in different manners that are more economical. Additionally, the Shi epoxidation has been shown to affect kinetic resolution of a limited selection of olefins.[20] This method is also not widely used, but is of mechanistic interest.
Epoxide openings
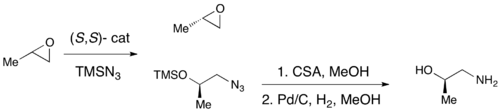
Jacobsen's (R,R) (salen)-Cr catalyst for kinetic resolution of terminal epoxides via azide formation
While enantioselective epoxidations have been successfully achieved utilizing Sharpless epoxidation, Shi epoxidation, and Jacobsen epoxidation, none of these methods allows for the efficient asymmetric synthesis of terminal epoxides, which are key chiral building blocks. Due to the inexpensiveness of most racemic terminal epoxides and their inability to generally be subjected to classical resolution, an effective kinetic resolution of terminal epoxides would serve as a highly important synthetic methodology. In 1996, Jacobsen and coworkers developed a methodology for the kinetic resolution of epoxides via nucleophilic ring-opening with attack by an azide anion. The (R,R) catalyst is shown.[21] The catalyst could effectively, with loadings as low as 0.5mol%, open the epoxide at the terminal position enantioselectively, yielding enantioenriched epoxide starting material and 1,2-azido alcohols. Yields are nearly quantitative and ee's were excellent (≥95% in nearly all cases). The 1,2-azido alcohols can be hydrogenated to give 1,2-amino alcohols, as shown below.
Jacobsen's (R,R) (salen)-Cr catalyst for hydrolytic kinetic resolution of terminal epoxides
In 1997, Jacobsen's group published a methodology which improved upon their earlier work, allowing for the use of water as the nucleophile in the epoxide opening. Utilizing a nearly identical catalyst, ee's in excess of 98% for both the recovered starting material epoxide and 1,2-diol product were observed. In the example below, hydrolytic kinetic resolution (HKR) was carried out on a 58 gram scale, resulting in 26 g (44%) of the enantioriched epoxide in >99% ee and 38 g (50%) of the diol in 98% ee.[22]
A multitude of other substrates were examined, with yields of the recovered epoxide ranging from 36-48% for >99% ee. Jacobsen hydrolytic kinetic resolution can be used in tandem with Jacobsen epoxidation to yield enantiopure epoxides from certain olefins, as shown below. The first epoxidation yields a slightly enantioenriched epoxide, and subsequent kinetic resolution yields essentially a single enantiomer. The advantage of this approach is the ability to reduce the amount of hydrolytic cleavage necessary to achieve high enantioselectivity, allowing for overall yields up to approximately 90%, based on the olefin.[23]
Ultimately, the Jacobsen epoxide opening kinetic resolutions produce high enantiomeric purity in the epoxide and product, in solvent-free or low-solvent conditions, and have been applicable on a large scale. The Jacobsen methodology for HKR in particular is extremely attractive since it can be carried out on a multiton scale and utilizes water as the nucleophile, resulting in extremely cost-effective industrial processes. Despite impressive achievements, HKR has generally been applied to the resolution of simple terminal epoxides with one stereocentre. Quite recently, D. A. Devalankar et al. reported an elegant protocol involving a two-stereocentered Co-catalyzed HKR of racemic terminal epoxides bearing adjacent C–C binding substituents.[24]
Oxidations
Noyori's (S,S) catalyst for the transfer hydrogenation/kinetic resolution of secondary alcohols
Ryōji Noyori and colleagues have developed a methodology for the kinetic resolution of benzylic and allylic secondary alcohols via transfer hydrogenation. The ruthenium complex catalyzes oxidation of the more reactive enantiomer from acetone, yielding an unreacted enantiopure alcohol, an oxidized ketone, and isopropanol. In the example illustrated below, exposure of 1-phenylethanol to the (S,S) enantiomer of the catalyst in the presence of acetone results in a 51% yield of 94% ee (R)-1-phenylethanol, along with 49% acetophenone and isopropanol as a byproduct.[25]
This methodology is essentially the reverse of Noyori's asymmetric transfer hydrogenation of ketones,[26] which yield enantioenriched alcohols via reduction. This limits the attractiveness of the kinetic resolution method, since there is a similar method to achieve the same products without the loss of half the material. Thus, the kinetic resolution would only be carried out in an instance for which the racemic alcohol was at least one half the price of the ketone or significantly easier to access.
Uemura and Hidai's catalyst for transfer hydrogenation/kinetic resolution of secondary alcohols
In addition, Uemura and Hidai have developed a ruthenium catalyst for the kinetic resolution oxidation of benzylic alcohols, yielding highly enantioenriched alcohols in good yields.[27] The complex can, like Noyori's catalyst, affect transfer hydrogenation between a ketone and isopropanol to give an enantioenriched alcohol as well as affect kinetic resolution of a racemic alcohol, giving enantiopure alcohol (>99% ee) and oxidized ketone, with acetone as the byproduct. It is highly effective at reducing ketones enantioselectively, giving most benzylic alcohols in >99% ee and can resolve a number of racemic benzylic alcohols to give high yields (up to 49%) of single enantiomers, as shown below. This method has the same disadvantages as the Noyori kinetic resolution, namely that the alcohols can also be accessed via reduction of the ketones enantioselectively. Additionally, only one enantiomer of the catalyst has been reported.
Hydrogenation
Noyori's (R)-BINAP Ru catalyst for the hydrogenative kinetic resolution of allylic alcohols
Noyori has also demonstrated the kinetic resolution of allylic alcohols by asymmetric hydrogenation of the olefin.[28] Utilizing the Ru[BINAP] complex, selective hydrogenation can give high ee's of the unsaturated alcohol in addition to the hydrogenated alcohol, as shown below. Thus, a second hydrogenation of the enantioenriched allylic alcohol remaining will give enantiomerically pure samples of both enantiomers of the saturated alcohol. Noyori has resolved a number of allylic alcohols with good to excellent yields and good to excellent ee's (up to >99%).
Ring closing metathesis
Hoveyda and Schrock's catalyst for ring closing metathesis kinetic resolution
Hoveyda and Schrock have developed a catalyst for ring-closing metathesis kinetic resolution of dienyl allylic alcohols.[29] The molybdenum alkylidene catalyst selectively catalyzes one enantiomer to perform ring closing metathesis, resulting in an enantiopure alcohol, and an enantiopure closed ring, as shown below. The catalyst is most effective at resolving 1,6-dienes. However, slight structural changes in the substrate, such as increasing the inter-alkene distance to 1,7, can sometimes necessitate the use of a different catalyst, reducing the efficacy of this method.
Enzymatic reactions
Acylations
As with synthetic kinetic resolution procedures, enzymatic acylation kinetic resolutions have seen the broadest application in a synthetic context. Especially important has been the use of enzymatic kinetic resolution to efficiently and cheaply prepare amino acids. On a commercial scale, Degussa's methodology employing acylases is capable of resolving numerous natural and unnatural amino acids. The racemic mixtures can be prepared via Strecker synthesis, and the use of either porcine kidney acylase (for straight chain substrates) or an enzyme from the mold Aspergillus oryzae (for branched side chain substrates) can effectively yield enantioenriched amino acids in high (85-90%) yields. The unreacted starting material can be racemized in situ, thus making this a dynamic kinetic resolution.[30]
In addition, lipases are used extensively for kinetic resolution in both academic and industrial settings.[31][32] Lipases have been used to resolve primary alcohols, secondary alcohols, a limited number of tertiary alcohols, carboxylic acids, diols, and even chiral allenes. Lipase from Pseudomonas cepacia (PSL) is the most widely used in the resolution of primary alcohols and has been used with vinyl acetate as an acylating agent to kinetically resolve the primary alcohols shown below.
For the resolution of secondary alcohols, pseudomonas cepecia lipase (PSL-C) has been employed effectively to generate excellent ee's of the (R)-enantiomer of the alcohol.[33] The use of isopropenyl acetate as the acylating agent results in acetone as the byproduct, which is effectively removed from the reaction using molecular sieves.
Oxidations and reductions
Baker's yeast (BY) has been utilized for the kinetic resolution of α-stereogenic carbonyl compounds.[34][35] The enzyme selectively reduces one enantiomer, yielding a highly enantioenriched alcohol and ketone, as shown below.
Baker's yeast has also been used in the kinetic resolution of secondary benzylic alcohols by oxidation.[36] While excellent ee's of the recovered alcohol have been reported, they typically require >60% conversion, resulting in diminished yields. Baker's yeast has also been used in the kinetic resolution via reduction of β-ketoesters.[37] However, given the success of Noyori's resolution of the same substrates, detailed later in this article, this has not seen much use.
Dynamic kinetic resolution (DKR) occurs when the starting material racemate is able to epimerize easily, resulting in an essentially racemic starting material mix at all points during the reaction. Then, the enantiomer with the lower barrier to activation can form in, theoretically, up to 100% yield. This is in contrast to standard kinetic resolution, which necessarily has a maximum yield of 50%. For this reason, dynamic kinetic resolution has extremely practical applications to organic synthesis. The observed dynamics are based on the Curtin-Hammett principle. The barrier to reaction of either enantiomer is necessarily higher than the barrier to epimerization, resulting in a kinetic well containing the racemate. This is equivalent to writing, for kR>kS,
A number of excellent reviews have been published, most recently in 2008, detailing the theory and practical applications of DKR.[38][39][40]
Noyori asymmetric hydrogenation
The Noyori asymmetric hydrogenation of ketones is an excellent example of dynamic kinetic resolution at work. The enantiomeric β-ketoesters can undergo epimerization, and the choice of chiral catalyst, typically of the form Ru[(R)-BINAP]X2, where X is a halogen, leads to one of the enantiomers reacting preferentially faster. The relative free energy for a representative reaction is shown below.[41][42] As can be seen, the epimerization intermediate is lower in free energy than the transition states for hydrogenation, resulting in rapid racemization and high yields of a single enantiomer of the product.
The enantiomers interconvert through their common enol, which is the energetic minimum located between the enantiomers. The shown reaction yields a 93% ee sample of the anti product shown above. Solvent choice appears to have a major influence on the diastereoselectivity, as dichloromethane and methanol both show effectiveness for certain substrates. Noyori and others have also developed newer catalysts which have improved on both ee and diastereomeric ratio (dr).
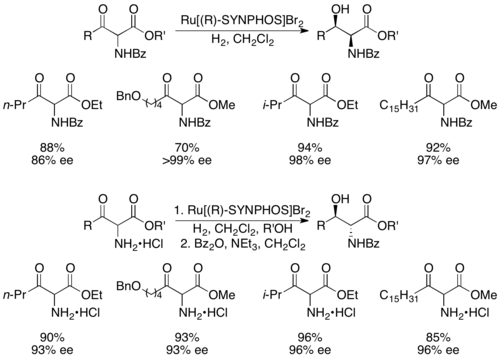
(S)-SYNPHOS
Genêt and coworkers developed SYNPHOS, a BINAP analogue which forms ruthenium complexes, which perform highly selective asymmetric hydrogenations.[43] Enantiopure Ru[SYNPHOS]Br2 was shown to selectively hydrogenate racemic α-amino-β-ketoesters to enantiopure aminoalcohols, as shown below utilizing (R)-SYNPHOS.[44] 1,2-syn amino alcohols were prepared from benzoyl protected amino compounds, whereas anti products were prepared from hydrochloride salts of the amine.
Fu acylation modification
Ruthenium catalyst used by Fu for racemization of secondary alcohols
Recently, Gregory Fu and colleagues reported a modification of their earlier kinetic resolution work to produce an effective dynamic kinetic resolution.[45] Using the ruthenium racemization catalyst shown to the right, and his planar chiral DMAP catalyst, Fu has demonstrated the dynamic kinetic resolution of secondary alcohols yielding up to 99% and 93% ee, as shown below. Work is ongoing to further develop the applications of the widely used DMAP catalyst to dynamic kinetic resolution.
Enzymatic dynamic kinetic resolutions
A number of enzymatic dynamic kinetic resolutions have been reported.[46] A prime example using PSL effectively resolves racemic acyloins in the presence of triethylamine and vinyl acetate as the acylating agent.[47] As shown below, the product was isolated in 75% yield and 97% ee. Without the presence of the base, regular kinetic resolution occurred, resulting in 45% yield of >99% ee acylated product and 53% of the starting material in 92% ee.
Another excellent, though not high-yielding, example is the kinetic resolution of (±)-8-amino-5,6,7,8-tetrahydroquinoline. When exposed to Candida antarctica lipase B (CALB) in toluene and ethyl acetate for 3–24 hours, normal kinetic resolution occurs, resulting in 45% yield of 97% ee of starting material and 45% yield of >97% ee acylated amine product. However, when the reaction is allowed to stir for 40–48 hours, racemic starting material and >60% of >95% ee acylated product are recovered.[48]
Here, the unreacted starting material racemizes in situ via a dimeric enamine, resulting in a recovery of greater than 50% yield of the enantiopure acylated amine product.
Chemoenzymatic dynamic kinetic resolutions
There have been a number of reported procedures which take advantage of a chemical reagent/catalyst to perform racemization of the starting material and an enzyme to selectively react with one enantiomer, called chemoenzymatic dynamic kinetic resolutions.[49] PSL-C was utilized along with a ruthenium catalyst (for racemization) to produce enantiopure (>95% ee) δ-hydroxylactones.[50]
Bäckvall's ruthenium catalyst for racemization in chemoenzymatic dynamic kinetic resolution of secondary alcohols
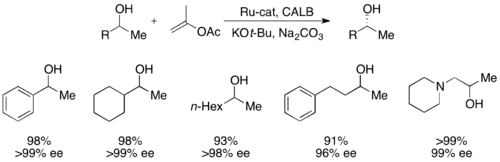
More recently, secondary alcohols have been resolved by Bäckvall with yields up to 99% and ee's up to >99% utilizing CALB and a ruthenium racemization complex.[51]
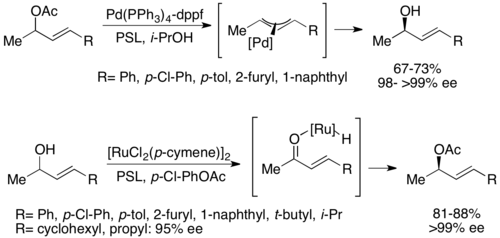
A second type of chemoenzymatic dynamic kinetic resolution involves a π-allyl complex from an allylic acetate with palladium. Here, racemization occurs with loss of the acetate, forming a cationic complex with the transition metal center, as shown below.[52] Palladium has been shown to facilitate this reaction, while ruthenium has been shown to affect a similar reaction, also shown below.[53]
Parallel kinetic resolution
In parallel kinetic resolution (PKR), a racemic mixture reacts to form two non-enantiomeric products, often through completely different reaction pathways. With PKR, there is no tradeoff between conversion and ee, as the formed products are not enantiomers.[54][55] One strategy for PKR is to remove the less reactive enantiomer (towards the desired chiral catalyst) from the reaction mixture by subjecting it to a second set of reaction conditions that preferentially react with it, ideally with an approximately equal reaction rate. Thus, both enantiomers are consumed in different pathways at equal rates. PKR experiments can be stereodivergent, regiodivergent, or structurally divergent.[56] One of the most highly efficient PKR's reported to date was accomplished by Yoshito Kishi in 1998; CBS reduction of a racemic steroidal ketone resulted in stereoselective reduction, producing two diastereomers of >99% ee, as shown below.[57]
PKR have also been accomplished with the use of enzyme catalysts. Using the fungus Mortierella isabellina NRRL 1757, reduction of racemic β-ketonitriles affords two diastereomers, which can be separated and re-oxidized to give highly enantiopure β-ketonitriles.[58] Highly synthetically useful parallel kinetic resolutions have truly yet to be discovered, however. A number of procedures have been discovered that give acceptable ee's and yields, but there are very few examples which give highly selective parallel kinetic resolution and not simply somewhat selective reactions. For example, Fu's parallel kinetic resolution of 4-alkynals yields very enantioenriched cyclobutanone in low yield and slightly enantioenriched cyclopentenone, as shown below.[59]
In theory, parallel kinetic resolution can give the highest ee's of products, since only one enantiomer gives each desired product. For example, for two complementary reactions both with s=49, 100% conversion would give products in 50% yield and 96% ee. These same values would require s=200 for a simple kinetic resolution. As such, the promise of PKR continues to attract much attention. The Kishi CBS reduction remains one of the few examples to fulfill this promise.
↑Fiaud, J.C.; Kagan, H.B. (1988). "Kinetic Resolution". In Eliel, E.L.; Wilen, S.H. (eds.). Topics in Stereochemistry. Vol.18. New York: John Wiley and Sons, Inc. pp.249–340.
↑Wurz, R.P.; Lee, E.C.; Ruble, J.C.; Fu, G.C. (2007). "Synthesis and Resolution of Planar-Chiral Derivatives of 4-(Dimethylamino)pyridine". Adv. Synth. Catal. 349 (14–15): 2345–2352. doi:10.1002/adsc.200700219.
↑Ruble, J.C.; Latham, H.A.; Fu, G.C. (1997). "Effective Kinetic Resolution of Secondary Alcohols with a Planar-Chiral Analogue of 4-(Dimethylamino)pyridine. Use of the Fe(C5Ph5) Group in Asymmetric Catalysis". J. Am. Chem. Soc. 119 (6): 1492–1493. doi:10.1021/ja963835b.
↑Ruble, J.C.; Tweddell, J.; Fu, G.C. (1998). "Kinetic Resolution of Arylakylcarbinols Catalyzed by a Planar-Chiral Derivative of DMAP: A New Benchmark for Nonenzymatic Acylation". J. Org. Chem. 63 (9): 2794–2795. doi:10.1021/jo980183w.
↑Tao, B.; Ruble, J.C.; Hoic, D.A.; Fu, G.C. (1999). "Nonenzymatic Kinetic Resolution of Propargylic Alcohols by a Planar−Chiral DMAP Derivative: Crystallographic Characterization of the Acylated Catalyst". J. Am. Chem. Soc. 121 (21): 2091–5092. Bibcode:1999JAChS.121.5091T. doi:10.1021/ja9906958.
↑Bellemin-Laponnaz, S.; Tweddell, J.; Ruble, J.C.; Breitling, F.M.; Fu, G.C. (2000). "The kinetic resolution of allylic alcohols by a non-enzymatic acylation catalyst; application to natural product synthesis". Chem. Commun. (12): 2091–5092. doi:10.1039/B002041I.
↑Martin, V.; Woodard, S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K.B. (1981). "Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity?". J. Am. Chem. Soc. 103 (23): 6237–6240. Bibcode:1981JAChS.103.6237M. doi:10.1021/ja00410a053.
↑Gao, Yun; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. (1987). "Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization". J. Am. Chem. Soc. 109 (19): 5765–5780. Bibcode:1987JAChS.109.5765G. doi:10.1021/ja00253a032.
↑Kitano, Y.; Matsumoto, T.; Sato, F. (1988). "A highly efficient kinetic resolution of γ- and β- trimethylsilyl secondary allylic alcohols by the sharpless asymmetric epoxidation". Tetrahedron. 44 (13): 4073–4086. doi:10.1016/S0040-4020(01)86657-6.
↑VanNieuwenhze, M.S.; Sharpless, K.B. (1993). "Kinetic resolution of racemic olefins via asymmetric dihydroxylation". J. Am. Chem. Soc. 115 (17): 7864–7865. Bibcode:1993JAChS.115.7864V. doi:10.1021/ja00070a037.
↑Corey, E.J.; Noe, M.C.; Guzman-Perez, A. (1995). "Kinetic Resolution by Enantioselective Dihydroxylation of Secondary Allylic 4-Methoxybenzoate Esters Using a Mechanistically Designed Cinchona Alkaloid Catalyst". J. Am. Chem. Soc. 117 (44): 10817–10824. Bibcode:1995JAChS.11710817C. doi:10.1021/ja00149a004.
↑Lorenz, J.C.; Frohn, M.; Zhou, X.; Zhang, J.-R.; Tang, Y.; Burke, C.; Shi, Y. (2005). "Transition State Studies on the Dioxirane-Mediated Asymmetric Epoxidation via Kinetic Resolution and Desymmetrization". J. Org. Chem. 70 (8): 2904–2911. doi:10.1021/jo048217p. PMID15822948.
↑Larrow, J.F.; Schaus, S.E.; Jacobsen, E.N. (1996). "Kinetic Resolution of Terminal Epoxides via Highly Regioselective and Enantioselective Ring Opening with TMSN3. An Efficient, Catalytic Route to 1,2-Amino Alcohols". J. Am. Chem. Soc. 118 (31): 7420–7421. Bibcode:1996JAChS.118.7420L. doi:10.1021/ja961708+.
↑Tokunaga, M.; Larrow, J.F.; Kakiuchi, F.; Jacobsen, E.N. (1997). "Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis". Science. 277 (5328): 936–938. doi:10.1126/science.277.5328.936. PMID9252321.
↑Brandes, B.D.; Jacobsen, E.N. (1997). "Synthesis of enantiopure 3-chlorostyrene oxide via an asymmetric epoxidation-hydrolytic kinetic resolution sequence". Tet. Asymm. 8 (23): 3927–3933. doi:10.1016/S0957-4166(97)00568-5.
↑Sudalai, A.; Karabal, P.U.; Devalankar, D.A. (2013). "Optically pure γ-butyrolactones and epoxy esters via two stereocentered HKR of 3-substituted epoxy esters: a formal synthesis of (−)-paroxetine, Ro 67-8867 and(+)-eldanolide". Org. Biomol. Chem. 11 (8): 1280–1285. doi:10.1039/c3ob27321k. PMID23334653.
↑Santaniello, E.; Ferraboschi, P.; Grisenti, P.; Manzocchi, A. (1992). "The biocatalytic approach to the preparation of enantiomerically pure chiral building blocks". Chem. Rev. 92 (5): 1071–1140. doi:10.1021/cr00013a016.
↑Ticozzi, C.; Zanarotti, Antonio (1989). "Enantioselective Microbial Reduction of 5-Acetylisoxazolines – A Novel Method for Stereochemical Control on Yeast Reduction". Liebigs Ann. Chem. 1989 (12): 1257–1259. doi:10.1002/jlac.198919890299.
↑Fantin, G.; Fogagnolo, M.; Medici, A.; Pedrini, P.; Poli, S. (1993). "Kinetic resolution of 1-aryl- and 1-heteroaryl ethanols by oxidation with Baker's yeast". Tetrahedron Lett. 34 (5): 883–884. doi:10.1016/0040-4039(93)89039-S. hdl:11392/462444.
↑Brooks, D.W.; Wilson, M.; Webb, M. (1987). "Different enzymic reactions of an enantiomeric pair: simultaneous dual kinetic resolution of a keto ester by bakers' yeast". J. Org. Chem. 52 (11): 2244–2248. doi:10.1021/jo00387a026.
↑Pellissier, H. (2008). "Recent developments in dynamic kinetic resolution". Tetrahedron. 64 (8): 1563–1601. doi:10.1016/j.tet.2007.10.080.
↑Kitamura, M.; Tokunaga, M.; Noyori, R. (1993). "Quantitative expression of dynamic kinetic resolution of chirally labile enantiomers: stereoselective hydrogenation of 2-substituted 3-oxo carboxylic esters catalyzed by BINAP-ruthenium(II) complexes". J. Am. Chem. Soc. 115 (1): 144–152. Bibcode:1993JAChS.115..144K. doi:10.1021/ja00054a020.
↑Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura, M.; Takaya, H.; Akutagawa, S.; Sayo, N.; Saito, T. (1989). "Stereoselective hydrogenation via dynamic kinetic resolution". J. Am. Chem. Soc. 111 (25): 9134–9135. Bibcode:1989JAChS.111.9134N. doi:10.1021/ja00207a038.
↑de Paule, S.D.; Jeulin, S.; Ratovelomanana-Vidal, V.; Genêt, J-P.; Champion, N.; Dellis, P. (2003). "Synthesis and Molecular Modeling Studies of SYNPHOS®, a New, Efficient Diphosphane Ligand For Ruthenium-Catalyzed Asymmetric Hydrogenation". Eur. J. Org. Chem. 2003 (10): 1931–1941. doi:10.1002/ejoc.200200634.
↑Mordant, C.; Ratovelomanana-Vidal, V.; Dünkelmann, P.; Genêt, J.-P. (2004). "A Versatile Route to syn- and anti-α-Amino β-Hydroxy Esters from β-Keto Esters by Dynamic Kinetic Resolution with Ru-SYNPHOS® Catalyst". Eur. J. Org. Chem. 2004 (14): 3017–3026. doi:10.1002/ejoc.200400078.
↑Pellissier, H. (2003). "Lipase–triethylamine-mediated dynamic transesterification of a tricyclic acyloin having a latent meso-structure: a new route to optically pure oxodicyclopentadiene". Tetrahedron. 59 (42): 8291–9327. doi:10.1016/S0040-4020(03)01022-6.
↑Taniguchi, T.; Ogasawara, K. (1997). "Lipase–triethylamine-mediated dynamic transesterification of a tricyclic acyloin having a latent meso-structure: a new route to optically pure oxodicyclopentadiene". Chemical Communications (15): 1399–1400. doi:10.1039/A702910A.
↑Pàmies, O.; Bäckvall, J.-E. (2002). "Enzymatic Kinetic Resolution and Chemoenzymatic Dynamic Kinetic Resolution of δ-Hydroxy Esters. An Efficient Route to Chiral δ-Lactones". J. Org. Chem. 67 (4): 1261–1265. doi:10.1021/jo016096c. PMID11846671.
↑Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F.B.; Bäckvall, J.-E. (2005). "Combined Ruthenium(II) and Lipase Catalysis for Efficient Dynamic Kinetic Resolution of Secondary Alcohols. Insight into the Racemization Mechanism". J. Am. Chem. Soc. 127 (64): 8817–8825. Bibcode:2005JAChS.127.8817M. doi:10.1021/ja051576x. PMID15954789.
↑Choi, Y.K.; Suh, J.H.; Lee, D.; Lim, I.T.; Jung, J.Y.; Kim, M.-J. (1999). "Dynamic Kinetic Resolution of Acyclic Allylic Acetates Using Lipase and Palladium". J. Org. Chem. 64 (22): 8423–8424. doi:10.1021/jo990956w. PMID11674772.
↑Lee, D.; Huh, E.A.; Kim, M.-J.; Jung, H.M.; Koh, J.H.; Park, J. (2000). "Dynamic Kinetic Resolution of Allylic Alcohols Mediated by Ruthenium- and Lipase-Based Catalysts". Org. Lett. 2 (15): 2377–2379. doi:10.1021/ol006159y. PMID10930288.
↑Dehil, J.R.; Gotor, V. (2002). "Parallel kinetic resolution of racemic mixtures: a new strategy for the preparation of enantiopure compounds?". Chem. Soc. Rev. 31 (6): 365–370. doi:10.1039/B205280F. PMID12491751.
↑Vedejs, E.; Jure, M. (2005). "Efficiency in Nonenzymatic Kinetic Resolution". Angew. Chem. Int. Ed. 44 (5): 3974–4001. doi:10.1002/anie.200460842. PMID15942973.
↑Kurosu, M.; Kishi, Y. (1998). "A Novel Example for Optical Resolution of Racemic Ketones Originating from Batrachotoxin Synthesis". J. Org. Chem. 63 (18): 6100–6101. doi:10.1021/jo981416m. PMID11672234.
↑Dehil, J.R.; Gotor, V. (2002). "Preparation of Enantiopure Ketones and Alcohols Containing a Quaternary Stereocenter through Parallel Kinetic Resolution of β-Keto Nitriles". J. Org. Chem. 67 (5): 1716–1718. doi:10.1021/jo011092t. PMID11871913.
↑Tanaka, K.; Fu, G.C. (2003). "Parallel Kinetic Resolution of 4-Alkynals Catalyzed by Rh(I)/Tol-BINAP: Synthesis of Enantioenriched Cyclobutanones and Cyclopentenones". J. Am. Chem. Soc. 125 (27): 8078–8079. Bibcode:2003JAChS.125.8078T. doi:10.1021/ja035489l. PMID12837058.
Further reading
Dynamic Kinetic Resolutions. A MacMillan Group Meeting. Jake Wiener Link
Dynamic Kinetic Resolution:A Powerful Approach to Asymmetric Synthesis. Erik Alexanian Supergroup Meeting March 30, 2005 Link
Dynamic Kinetic Resolution: Practical Applications in Synthesis. Valerie Keller 3rd-Year Seminar November 1, 2001 Link
Kinetic Resolution. David Ebner Stoltz Group Literature Seminar. June 4, 2003 link
Kinetic Resolutions. UT Southwestern Presentation. link
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.