
The oxytocin receptor, also known as OXTR, is a protein which functions as receptor for the hormone and neurotransmitter oxytocin. In humans, the oxytocin receptor is encoded by the OXTR gene which has been localized to human chromosome 3p25.

Orexin receptor type 2 (Ox2R or OX2), also known as hypocretin receptor type 2 (HcrtR2), is a protein that in humans is encoded by the HCRTR2 gene. It should not be confused for the protein CD200R1 which shares the alias OX2R but is a distinct, unrelated gene located on the human chromosome 3.

Himbacine is an alkaloid isolated from the bark of Australian magnolias. Himbacine has been synthesized using a Diels-Alder reaction as a key step. Himbacine's activity as a muscarinic receptor antagonist, with specificity for the muscarinic acetylcholine receptor M2, made it a promising starting point in Alzheimer's disease research. The development of a muscarinic antagonist based on himbacine failed but an analog, vorapaxar, has been approved by the FDA as a thrombin receptor antagonist.

A-349,821 is a potent and selective histamine H3 receptor antagonist (or possibly an inverse agonist). It has nootropic effects in animal studies, although there do not appear to be any plans for clinical development at present and it is currently only used in laboratory research.

SB-357134 is a drug which is used in scientific research. It acts as a potent, selective and orally active 5-HT6 receptor antagonist. SB-357134 and other 5-HT6 antagonists show nootropic effects in animal studies, and have been proposed as potential novel treatments for cognitive disorders such as schizophrenia and Alzheimer's disease.
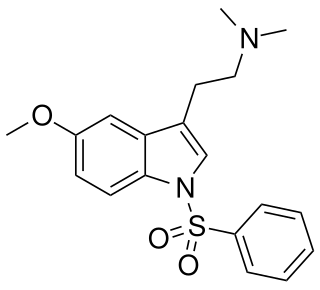
MS-245 is a tryptamine derivative used in scientific research. It acts as a selective 5-HT6 receptor antagonist with a Ki of 2.3 nM, and was derived through structure-activity relationship development of the selective 5-HT6 agonist EMDT. It has been used as a lead compound for further development of tryptamine-derived 5-HT6 antagonists. In animal studies it has been shown to boost the activity of, but not substitute for, both amphetamine and nicotine.

J-113,397 is an opioid drug which was the first compound found to be a highly selective antagonist for the nociceptin receptor, also known as the ORL-1 receptor. It is several hundred times selective for the ORL-1 receptor over other opioid receptors, and its effects in animals include preventing the development of tolerance to morphine, the prevention of hyperalgesia induced by intracerebroventricular administration of nociceptin, as well as the stimulation of dopamine release in the striatum, which increases the rewarding effects of cocaine, but may have clinical application in the treatment of Parkinson's disease.

BIBP-3226 is a drug used in scientific research which acts as a potent and selective antagonist for both the Neuropeptide Y receptor Y1 and also the neuropeptide FF receptor. It was the first non-peptide antagonist developed for the Y1 receptor and has been widely used to help determine its functions in the body. Activation of Y1 is thought to be involved in functions such as regulation of appetite and anxiety, and BIBP-3226 has anxiogenic and anorectic effects, as well as blocking the Y1-mediated corticotropin releasing hormone release. It has also been used as a lead compound to develop a number of newer more potent Y1 antagonists.

CECXG (3'-ethyl-LY-341,495) is a research drug which acts as a potent and selective antagonist for the group II metabotropic glutamate receptors (mGluR2/3), with reasonable selectivity for mGluR3. While it is some five times less potent than LY-341,495 at mGluR3, it has 38x higher affinity for mGluR3 over mGluR2, making it one of the few ligands available that is able to distinguish between these two closely related receptor subtypes.

CSP-2503 is a potent and selective 5-HT1A receptor agonist, 5-HT2A receptor antagonist, and 5-HT3 receptor antagonist of the naphthylpiperazine class. First synthesized in 2003, it was designed based on computational models and QSAR studies. In rat studies, CSP-2503 has demonstrated anxiolytic effects, and thus has been suggested as a treatment for anxiety in humans with a multimodal mechanism of action.

L-368,899 is a drug used in scientific research which acts as a selective antagonist of the oxytocin receptor, with good selectivity over the related vasopressin receptors. Unlike related drugs such as the peripherally selective L-371,257, the oral bioavailabity is high and the brain penetration of L-368,899 is rapid, with selective accumulation in areas of the limbic system. This makes it a useful tool for investigating the centrally mediated roles of oxytocin, such as in social behaviour and pair bonding, and studies in primates have shown L-368,899 to reduce a number of behaviours such as food sharing, sexual activity and caring for infants, demonstrating the importance of oxytocinergic signalling in mediating these important social behaviours.
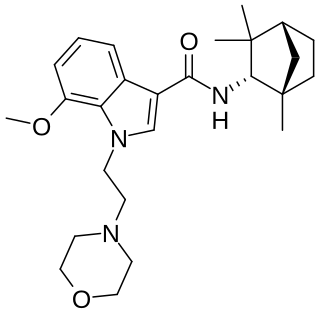
MN-25 (UR-12) is a drug invented by Bristol-Myers Squibb, that acts as a reasonably selective agonist of peripheral cannabinoid receptors. It has moderate affinity for CB2 receptors with a Ki of 11 nM, but 22x lower affinity for the psychoactive CB1 receptors with a Ki of 245 nM. The indole 2-methyl derivative has the ratio of affinities reversed however, with a Ki of 8 nM at CB1 and 29 nM at CB2, which contrasts with the usual trend of 2-methyl derivatives having increased selectivity for CB2 (cf. JWH-018 vs JWH-007, JWH-081 vs JWH-098).

Epelsiban is an orally bioavailable drug which acts as a selective and potent oxytocin receptor antagonist. It was initially developed by GlaxoSmithKline (GSK) for the treatment of premature ejaculation in men and then as an agent to enhance embryo or blastocyst implantation in women undergoing embryo or blastocyst transfer associated with in vitro fertilization (IVF)., and was also investigated for use in the treatment of adenomyosis.

Biphalin is a dimeric enkephalin endogenous peptide (Tyr-D-Ala-Gly-Phe-NH)2 composed of two tetrapeptides derived from enkephalins, connected 'tail-to-tail' by a hydrazide bridge. The presence of two distinct pharmacophores confers on biphalin a high affinity for both μ and δ opioid receptors (with an EC50 of about 1–5 nM for both μ and δ receptors), therefore it has analgesic activity. Biphalin presents a considerable antinociceptive profile. In fact, when administered intracerebroventricularly in mice, biphalin displays a potency almost 7-fold greater than that of the ultra-potent alkaloid agonist, etorphine and 7000-fold greater than morphine; biphalin and morphine were found to be equipotent after intraperitoneal administration. The extraordinary in vivo potency shown by this compound is coupled with low side-effects, in particular, to produce no dependency in chronic use. For these reasons, several efforts have been carried out in order to obtain more information about structure-activity relationship (SAR). Results clearly indicate that, at least for μ receptor binding, the presence of two pharmacophores is not necessary; Tyr1 is indispensable for analgesic activity, while replacing Phe at the position 4 and 4' with non-aromatic, but lipophilic amino acids does not greatly change the binding properties and in general 4,4' positions are found to be important to design biphalin analogues with increased potency and modified μ/δ selectivity. The hydrazide linker is not fundamental for activity or binding, and it can be conveniently substituted by different conformationally constrained cycloaliphatic diamine linkers.
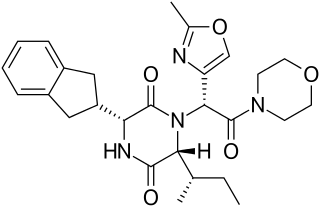
Retosiban also known as GSK-221,149-A is an oral drug which acts as an oxytocin receptor antagonist. It is being developed by GlaxoSmithKline for the treatment of preterm labour. Retosiban has high affinity for the oxytocin receptor and has greater than 1400-fold selectivity over the related vasopressin receptors

KM-233 is a synthetic cannabinoid drug which is a structural analog of Δ8-tetrahydrocannabinol (THC), the less active but more stable isomer of the active component of Cannabis. KM-233 differs from Δ8-THC by the pentyl side chain being replaced by a 1,1-dimethylbenzyl group. It has high binding affinity in vitro for both the CB1 and CB2 receptors, with a CB2 affinity of 0.91 nM and 13-fold selectivity over the CB1 receptor. In animal studies, it has been found to be a potential treatment for glioma, a form of brain tumor. Many related analogues are known where the 1,1-dimethylbenzyl group is substituted or replaced by other groups, with a fairly well established structure-activity relationship.

TC OT 39 is a non-peptide partial agonist of the oxytocin and vasopressin V2 receptors (Ki = 147 nM and >1000 nM, respectively) and antagonist of the vasopressin V1A receptor (Ki = 330 nM).
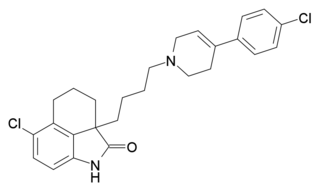
DR-4485 is a compound which acts as a potent and selective antagonist for the 5-HT7 receptor, with good oral bioavailability. It has been used to research the function of this still comparatively little studied serotonin receptor subtype.

Sufugolix (INNTooltip International Nonproprietary Name, BANTooltip British Approved Name) (developmental code name TAK-013) is a non-peptide, orally-active, selective antagonist of the gonadotropin-releasing hormone receptor (GnRHR) (IC50Tooltip Half-maximal inhibitory concentration = 0.1 and 0.06 nM for affinity and in vitro inhibition, respectively). It was under development by Takeda for the treatment of endometriosis and uterine leiomyoma and reached phase II clinical trials for both of these indications, but was subsequently discontinued. It seems to have been supplanted by relugolix (TAK-385), which is also under development by Takeda for the treatment of these conditions and has a more favorable drug profile (including reduced cytochrome P450 inhibition and improved in vivo GnRHR antagonistic activity) in comparison.

Emma Parmee is a British chemist and research scientist who is a co-inventor of numerous drug patents. She was one of the leading researchers in the development of sitagliptin and was awarded a Thomas Alva Edison Patent Award in 2007 and the Society of Chemical Industry's Gordon E Moore Medal in 2009 for her contributions.
