
Neurofibromatosis (NF) is a group of three conditions in which tumors grow in the nervous system. The three types are neurofibromatosis type I (NF1), neurofibromatosis type II (NF2), and schwannomatosis. In NF1 symptoms include light brown spots on the skin, freckles in the armpit and groin, small bumps within nerves, and scoliosis. In NF2, there may be hearing loss, cataracts at a young age, balance problems, flesh colored skin flaps, and muscle wasting. In schwannomatosis there may be pain either in one location or in wide areas of the body. The tumors in NF are generally non-cancerous.

A soft-tissue sarcoma (STS) is a malignant tumor, a type of cancer, that develops in soft tissue. A soft-tissue sarcoma is often a painless mass that grows slowly over months or years. They may be superficial or deep-seated. Any such unexplained mass must be diagnosed by biopsy. Treatment may include surgery, radiotherapy, chemotherapy, and targeted drug therapy. Bone sarcomas are the other class of sarcomas.

A sarcoma is a malignant tumor, a type of cancer that arises from cells of mesenchymal origin. Connective tissue is a broad term that includes bone, cartilage, fat, vascular, or other structural tissues, and sarcomas can arise in any of these types of tissues. As a result, there are many subtypes of sarcoma, which are classified based on the specific tissue and type of cell from which the tumor originates. Sarcomas are primary connective tissue tumors, meaning that they arise in connective tissues. This is in contrast to secondary connective tissue tumors, which occur when a cancer from elsewhere in the body spreads to the connective tissue. Sarcomas are one of five different types of cancer, classified by the cell type from which they originate. The word sarcoma is derived from the Greek σάρκωμα sarkōma 'fleshy excrescence or substance', itself from σάρξsarx meaning 'flesh'.

A bone tumor is an abnormal growth of tissue in bone, traditionally classified as noncancerous (benign) or cancerous (malignant). Cancerous bone tumors usually originate from a cancer in another part of the body such as from lung, breast, thyroid, kidney and prostate. There may be a lump, pain, or neurological signs from pressure. A bone tumor might present with a pathologic fracture. Other symptoms may include fatigue, fever, weight loss, anemia and nausea. Sometimes there are no symptoms and the tumour is found when investigating another problem.

An osteosarcoma (OS) or osteogenic sarcoma (OGS) is a cancerous tumor in a bone. Specifically, it is an aggressive malignant neoplasm that arises from primitive transformed cells of mesenchymal origin and that exhibits osteoblastic differentiation and produces malignant osteoid.
Spinal tumors are neoplasms located in either the vertebral column or the spinal cord. There are three main types of spinal tumors classified based on their location: extradural and intradural. Extradural tumors are located outside the dura mater lining and are most commonly metastatic. Intradural tumors are located inside the dura mater lining and are further subdivided into intramedullary and extramedullary tumors. Intradural-intramedullary tumors are located within the dura and spinal cord parenchyma, while intradural-extramedullary tumors are located within the dura but outside the spinal cord parenchyma. The most common presenting symptom of spinal tumors is nocturnal back pain. Other common symptoms include muscle weakness, sensory loss, and difficulty walking. Loss of bowel and bladder control may occur during the later stages of the disease.

Fibrosarcoma is a malignant mesenchymal tumour derived from fibrous connective tissue and characterized by the presence of immature proliferating fibroblasts or undifferentiated anaplastic spindle cells in a storiform pattern. Fibrosarcomas mainly arise in people between the ages of 25 and 79. It originates in fibrous tissues of the bone and invades long or flat bones such as the femur, tibia, and mandible. It also involves the periosteum and overlying muscle.

Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of neurofibromin 1 (NF-1), a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.

A synovial sarcoma is a rare form of cancer which occurs primarily in the extremities of the arms or legs, often in proximity to joint capsules and tendon sheaths. It is a type of soft-tissue sarcoma.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.

A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
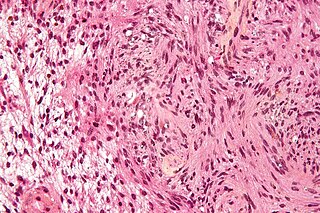
Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.
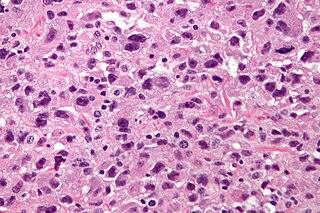
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

A schwannoma is a usually benign nerve sheath tumor composed of Schwann cells, which normally produce the insulating myelin sheath covering peripheral nerves.

Neurofibromin 1 (NF1) is a gene in humans that is located on chromosome 17. NF1 codes for neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations in NF1 can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.
A nerve sheath tumor is a type of tumor of the nervous system which is made up primarily of the myelin surrounding nerves. From benign tumors like schwannoma to high grade malignant neoplasms known as malignant peripheral nerve sheath tumors, peripheral nerve sheath tumors include a range of clearly characterized clinicopathologic entities. A peripheral nerve sheath tumor (PNST) is a nerve sheath tumor in the peripheral nervous system. Benign peripheral nerve sheath tumors include schwannomas and neurofibromas.
Soft tissue sarcoma refers to a broad group of tumors that originate from connective tissues. They tend to have similar histologic appearance and biological behavior, and can be either benign or malignant. Soft tissue sarcomas can arise in any part of the pet's body but skin and subcutaneous tumors are the most commonly observed. Soft-tissue sarcomas comprise approximately 15% of all skin and subcutaneous tumors in dogs and approximately 7% of all skin and subcutaneous tumors in cats. The variety of different tumors that fall under the category of soft tissue sarcomas includes fibrosarcoma, hemangiopericytoma, liposarcoma, rhabdomyosarcoma, leiomyosarcoma, malignant fibrous histiocytoma, malignant nerve sheath tumors, myxosarcoma, myxofibrosarcoma, mesenchymoma, and spindle cell tumor.
Malignant triton tumor (MTT) is a relatively rare, aggressive tumor made up of both malignant schwannoma cells and malignant rhabdomyoblasts. It is classified as a malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation.
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
Myxofibrosarcoma (MFS), although a rare type of tumor, is one of the most common soft tissue sarcomas, i.e. cancerous tumors, that develop in the soft tissues of elderly individuals. Initially considered to be a type of histiocytoma termed fibrous histiocytoma or myxoid variant of malignant fibrous histiocytoma, Angervall et al. termed this tumor myxofibrosarcoma in 1977. In 2020, the World Health Organization reclassified MFS as a separate and distinct tumor in the category of malignant fibroblastic and myofibroblastic tumors.



