
Androgen insensitivity syndrome (AIS) is a difference in sex development involving hormonal resistance due to androgen receptor dysfunction.

Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.

Thyroid hormone resistance (also resistance to thyroid hormone (RTH), and sometimes Refetoff syndrome) describes a rare syndrome in which the thyroid hormone levels are elevated but the thyroid stimulating hormone (TSH) level is not suppressed, or not completely suppressed as would be expected. The first report of the condition appeared in 1967. Essentially this is decreased end organ responsiveness to thyroid hormones. A new term "impaired sensitivity to thyroid hormone" has been suggested in March 2014 by Refetoff et al.
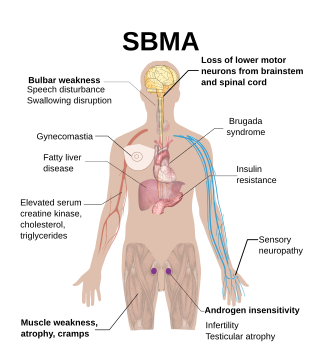
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.
Trinucleotide repeat disorders, a subset of microsatellite expansion diseases, are a set of over 30 genetic disorders caused by trinucleotide repeat expansion, a kind of mutation in which repeats of three nucleotides increase in copy numbers until they cross a threshold above which they cause developmental, neurological or neuromuscular disorders. Depending on its location, the unstable trinucleotide repeat may cause defects in a protein encoded by a gene; change the regulation of gene expression; produce a toxic RNA, or lead to production of a toxic protein. In general, the larger the expansion the faster the onset of disease, and the more severe the disease becomes.
Isolated hypogonadotropic hypogonadism (IHH), also called idiopathic or congenital hypogonadotropic hypogonadism (CHH), as well as isolated or congenital gonadotropin-releasing hormone deficiency (IGD), is a condition which results in a small subset of cases of hypogonadotropic hypogonadism (HH) due to deficiency in or insensitivity to gonadotropin-releasing hormone (GnRH) where the function and anatomy of the anterior pituitary is otherwise normal and secondary causes of HH are not present.

Corticosteroid 11-β-dehydrogenase isozyme 2 also known as 11-β-hydroxysteroid dehydrogenase 2 is an enzyme that in humans is encoded by the HSD11B2 gene.
Pseudohypoaldosteronism (PHA) is a condition that mimics hypoaldosteronism. However, the condition is due to a failure of response to aldosterone, and levels of aldosterone are actually elevated, due to a lack of feedback inhibition.
Partial androgen insensitivity syndrome (PAIS) is a condition that results in the partial inability of the cell to respond to androgens. It is an X linked recessive condition. The partial unresponsiveness of the cell to the presence of androgenic hormones impairs the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development. As such, the insensitivity to androgens is clinically significant only when it occurs in individuals with a Y chromosome. Clinical features include ambiguous genitalia at birth and primary amenhorrhoea with clitoromegaly with inguinal masses. Müllerian structures are not present in the individual.

Estrogen insensitivity syndrome (EIS), or estrogen resistance, is a form of congenital estrogen deficiency or hypoestrogenism which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. Congenital estrogen deficiency can alternatively be caused by a defect in aromatase, the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS.

Gonadotropin-releasing hormone receptor is a protein that in humans is encoded by the GNRHR gene.

The human gene SRD5A2 encodes the 3-oxo-5α-steroid 4-dehydrogenase 2 enzyme, also known as 5α-reductase type 2 (5αR2), one of three isozymes of 5α-reductase.

Thyroid hormone receptor beta (TR-beta) also known as nuclear receptor subfamily 1, group A, member 2 (NR1A2), is a nuclear receptor protein that in humans is encoded by the THRB gene.

Homeobox protein prophet of PIT-1 is a protein that in humans is encoded by the PROP1 gene.

Luteinizing hormone subunit beta also known as lutropin subunit beta or LHβ is a polypeptide that in association with an alpha subunit common to all gonadotropin hormones forms the reproductive signaling molecule luteinizing hormone. In humans it is encoded by the LHB gene.

Thyroid stimulating hormone, beta also known as TSHB is a protein which in humans is encoded by the TSHB gene.

Aromatase deficiency is a rare condition characterized by extremely low levels or complete absence of the enzyme aromatase activity in the body. It is an autosomal recessive disease resulting from various mutations of gene CYP19 (P450arom) which can lead to ambiguous genitalia and delayed puberty in females, continued linear growth into adulthood and osteoporosis in males and virilization in pregnant mothers. As of 2020, fewer than 15 cases have been identified in genetically male individuals and at least 30 cases in genetically female individuals.
Complete androgen insensitivity syndrome (CAIS) is an AIS condition that results in the complete inability of the cell to respond to androgens. As such, the insensitivity to androgens is only clinically significant when it occurs in individuals who are exposed to significant amounts of testosterone at some point in their lives. The unresponsiveness of the cell to the presence of androgenic hormones prevents the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does allow, without significant impairment, female genital and sexual development in those with the condition.
Kowarski syndrome describes cases of growth failure, despite the presence of normal or slightly high blood growth hormone by radioimmunoassay (RIA-GH) and low serum IGF1, and who exhibit a significant increase in growth rate following recombinant GH therapy.
Cytochrome P450 family 19 subfamily A member 1 is a protein that in humans is encoded by the CYP19A1 gene.


