
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

ICF syndrome is a very rare autosomal recessive immune disorder.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the development of the corpus callosum, the band of white matter connecting the two hemispheres in the brain, in the embryo is disrupted. The result of this is that the fibers that would otherwise form the corpus callosum are instead longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.
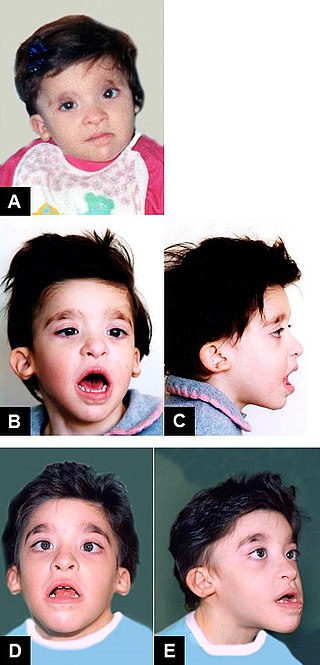
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000–100,000 births.

Kaufman oculocerebrofacial syndrome, also known as blepharophimosis-ptosis-intellectual disability syndrome, is an extremely rare autosomal recessive congenital disorder characterized by severe mental retardation, brachycephaly, upslanting palpebral fissures, eye abnormalities, and highly arched palate. It was characterized in 1971; eight cases had been identified as of 1995. To date, the amount of cases is disputed, with sources claiming the number ranges from 14 to 31.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

Probable E3 ubiquitin-protein ligase HERC1 is an enzyme that in humans is encoded by the HERC1 gene.

Acrocallosal syndrome is an extremely rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979. Mutations in KIF7 are causative for ACLS, and mutations in GLI3 are associated with a similar syndrome.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt–Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.
Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.
Genitopatellar syndrome is a rare disorder consisting of congenital flexion contractures of the lower extremities, abnormal or missing patellae, and urogenital anomalies. Additional symptoms include microcephaly, severe psychomotor disability. In 2012, it was shown that mutations in the gene KAT6B cause the syndrome. Genitopatellar syndrome (GTPTS) can be caused by heterozygous mutation in the KAT6B gene on chromosome 10q22. The Say-Barber-Biesecker variant of Ohdo syndrome, which has many overlapping features with GTPTS, can also be caused by heterozygous mutation in the KAT6B gene.

Lateral meningocele syndrome, also known as Lehman syndrome, is a very rare skeletal disorder with facial anomalies, hypotonia, and meningocele-related neurologic dysfunction. These protrusions form from membranes surrounding the spinal cord in gaps in the spine (vertebrae). They most often occur in the lower spine and damage the surrounding nerves that spread throughout the rest of the body. Examples of resulting damages are bladder function, prickling or tingling sensations, stiffness and weakness in the legs, and back pain. People affected with lateral meningocele typically have high arched eyebrows, widely spaced eyes, droopy eyes, and other facial features. There have been only 14 reported individuals with lateral meningocele syndrome with 7 of those who have a molecularly confirmed diagnosis. There is no specific treatment for this syndrome, but only supportive management including lateral spinal meningoceles, psychomotor development, musculoskeletal, and routine management.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
Bainbridge–Ropers syndrome was first identified in 2013 and is characterized by failure to thrive, feeding problems, hypotonia, intellectual disabilities, autism, postnatal growth delay, abnormal facial features such as arched eyebrows, anteverted nares, and delays in language acquisition. BRPS is extremely rare worldwide; more than thirty cases of BRPS have been reported abroad, and four cases have been reported in China.
Alwadei syndrome or autosomal recessive mental retardation-61 (MRT61) is an autosomal recessive neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, and variable abnormal facial features. Severe patients may develop refractory seizures and have brain abnormalities, including hypoplasia of the corpus callosum. Alwadei syndrome attributed to mutation in RUSC2 gene on chromosome 9p13.3.
DeSanto-Shinawi (DESSH) syndrome is a rare genetic disorder caused by genetic variations (mutations) in a gene called WW Domain-Containing Adaptor with Coiled-coil Region. The condition was first described in 2015 in six individuals. The prevalence of DESSH syndrome is unknown at this time, but 25 individuals have been so far described in the medical literature. However, many other individuals with this condition are being studied and characterized. With the increasing utilization of exome and whole genome sequencing, it is anticipated that many more individuals to be identified.
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
Multiple congenital anomalies-hypotonia-seizures syndrome is a rare multi-systemic genetic disorder which is characterized by developmental delay, seizures, hypotonia and heart, urinary, and gastrointestinal abnormalities.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
