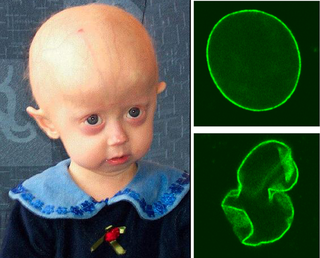
Progeria is a specific type of progeroid syndrome, also known as Hutchinson–Gilford syndrome or Hutchinson–Gilford progeroid syndrome (HGPS). A single gene mutation is responsible for causing progeria. The gene, known as lamin A (LMNA), makes a protein necessary for holding the nucleus of the cell together. When this gene gets mutated, an abnormal form of lamin A protein called progerin is produced. Progeroid syndromes are a group of diseases that cause individuals to age faster than usual, leading to them appearing older than they actually are. People born with progeria typically live until their mid to late teens or early twenties.

Lamins, also known as nuclear lamins are fibrous proteins in type V intermediate filaments, providing structural function and transcriptional regulation in the cell nucleus. Nuclear lamins interact with inner nuclear membrane proteins to form the nuclear lamina on the interior of the nuclear envelope. Lamins have elastic and mechanosensitive properties, and can alter gene regulation in a feedback response to mechanical cues. Lamins are present in all animals but are not found in microorganisms, plants or fungi. Lamin proteins are involved in the disassembling and reforming of the nuclear envelope during mitosis, the positioning of nuclear pores, and programmed cell death. Mutations in lamin genes can result in several genetic laminopathies, which may be life-threatening.
In genetics, a missense mutation is a point mutation in which a single nucleotide change results in a codon that codes for a different amino acid. It is a type of nonsynonymous substitution.
Malouf syndrome is a congenital disorder that causes one or more of the following symptoms: mental retardation, ovarian dysgenesis, congestive cardiomyopathy, broad nasal base, blepharoptosis, and bone abnormalities, and occasionally marfanoid habitus.

Emery–Dreifuss muscular dystrophy (EDMD) is a type of muscular dystrophy, a group of heritable diseases that cause progressive impairment of muscles. EDMD affects muscles used for movement, causing atrophy, weakness and contractures. It almost always affects the heart, causing abnormal rhythms, heart failure, or sudden cardiac death. It is rare, affecting 0.39 per 100,000 people. It is named after Alan Eglin H. Emery and Fritz E. Dreifuss.

Emerin is a protein that in humans is encoded by the EMD gene, also known as the STA gene. Emerin, together with LEMD3, is a LEM domain-containing integral protein of the inner nuclear membrane in vertebrates. Emerin is highly expressed in cardiac and skeletal muscle. In cardiac muscle, emerin localizes to adherens junctions within intercalated discs where it appears to function in mechanotransduction of cellular strain and in beta-catenin signaling. Mutations in emerin cause X-linked recessive Emery–Dreifuss muscular dystrophy, cardiac conduction abnormalities and dilated cardiomyopathy.

Prelamin-A/C, or lamin A/C is a protein that in humans is encoded by the LMNA gene. Lamin A/C belongs to the lamin family of proteins.

Restrictive dermopathy (RD) is a rare, lethal autosomal recessive skin condition characterized by syndromic facies, tight skin, sparse or absent eyelashes, and secondary joint changes.

Polyadenylate-binding protein 2 (PABP-2) also known as polyadenylate-binding nuclear protein 1 (PABPN1) is a protein that in humans is encoded by the PABPN1 gene. PABN1 is a member of a larger family of poly(A)-binding proteins in the human genome.

Delta-sarcoglycan is a protein that in humans is encoded by the SGCD gene.

Gamma-sarcoglycan is a protein that in humans is encoded by the SGCG gene. The α to δ-sarcoglycans are expressed predominantly (β) or exclusively in striated muscle. A mutation in any of the sarcoglycan genes may lead to a secondary deficiency of the other sarcoglycan proteins, presumably due to destabilisation of the sarcoglycan complex. The disease-causing mutations in the α to δ genes cause disruptions within the dystrophin-associated protein (DAP) complex in the muscle cell membrane. The transmembrane components of the DAP complex link the cytoskeleton to the extracellular matrix in adult muscle fibres, and are essential for the preservation of the integrity of the muscle cell membrane.

Torsin-1A-interacting protein 1 is a protein that in humans is encoded by the TOR1AIP1 gene. More commonly known as lamina associated polypeptide 1 (LAP1), it is a type II integral membrane protein that resides in the inner nuclear membrane. The luminal domain of LAP1 interacts with Torsin A and is necessary for the ATPase activity of Torsin A. LAP1 plays a critical role in skeletal and heart muscle. Mutations in TOR1AIP1 have been linked to muscular dystrophy and cardiomyopathy. It's deletion from mouse hepatocytes leads to defected very-low density lipoprotein secretion and causes non-alcoholic fatty liver disease and non-alcoholic steatohepatitis
ZMPSTE24 is a human gene. The protein encoded by this gene is a metallopeptidase. It is involved in the processing of lamin A. Defects in the ZMPSTE24 gene lead to similar laminopathies as defects in lamin A, because the latter is a substrate for the former. In humans, a mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. Failure to correctly process prelamin A leads to deficient ability to repair DNA double-strand breaks.

Progerin is a truncated version of the lamin A protein involved in the pathology of Hutchinson–Gilford progeria syndrome. Progerin is most often generated by a sporadic single point nucleotide polymorphism c.1824 C>T in the gene that codes for matured Lamin A. This mutation activates a cryptic splice site that induces a mutation in premature Lamin A with the deletion of a 50 amino acids group near the C-terminus. The endopeptidase ZMPSTE24 cannot cleave between the missing RSY - LLG amino acid sequence during the maturation of Lamin A, due to the deletion of the 50 amino acids which included that sequence. This leaves the intact premature Lamin A bonded to the methylated carboxyl farnesyl group creating the defective protein Progerin, rather than the desired protein matured Lamin A. Approximately 90% of all Hutchinson–Gilford progeria syndrome cases are heterozygous for this deleterious single nucleotide polymorphism within exon 11 of the LMNA gene causing the post-translational modifications to produce Progerin.
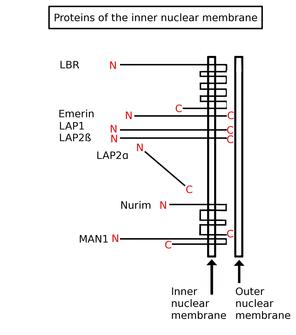
Inner nuclear membrane proteins are membrane proteins that are embedded in or associated with the inner membrane of the nuclear envelope. There are about 60 INM proteins, most of which are poorly characterized with respect to structure and function. Among the few well-characterized INM proteins are lamin B receptor (LBR), lamina-associated polypeptide 1 (LAP1), lamina-associated polypeptide-2 (LAP2), emerin and MAN1.
Mandibuloacral dysplasia (MAD) is a rare autosomal recessive syndrome characterized by mandibular hypoplasia, delayed cranial suture closure, dysplastic clavicles, abbreviated and club-shaped terminal phalanges, acroosteolysis, atrophy of the skin of the hands and feet, and typical facial changes.
Familial partial lipodystrophy, also known as Köbberling–Dunnigan syndrome, is a rare genetic metabolic condition characterized by the loss of subcutaneous fat.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
Veena Krishnaji Parnaik is an Indian cell biologist and the current Chief Scientist at the Centre for Cellular and Molecular Biology. She obtained her Masters in Science in medicinal biochemistry from the University of Mumbai and received her PhD from Ohio State University before moving back to India to work at the CCMB. Her research is focused on understanding the functional role of the nuclear lamina and how defects in it may lead to disorders such as progeria and muscular dystrophy.

Lamin A/C congenital muscular dystrophy (CMD) is a disease that it is included in laminopathies. Laminopathies are caused, among other mutations, to mutations in LMNA, a gene that synthesizes lamins A and C.
