Organic compound made by removing substituents from an aromatic ring
In organic chemistry, arynes[1] and benzynes[2] are a class of highly reactive chemical species derived from an aromatic ring by removal of two substituents. Arynes are examples of didehydroarenes (1,2-didehydroarenes in this case), although 1,3- and 1,4-didehydroarenes are also known.[3][4][5] Arynes are examples of alkynes under high strain.
The alkyne representation of benzyne is the most widely encountered. Arynes are usually described as having a strained triple bond (left), but resonance contributors include a cumulene form (middle) and biradical form (right):[6]
Geometric constraints on the triple bond in benzyne result in diminished overlap of in-plane p-orbitals, and thus weaker triple bond.[7] The vibrational frequency of the triple bond in benzyne was assigned by Radziszewski to be 1846cm−1,[8] indicating a weaker triple bond than in unstrained alkyne with vibrational frequency of approximately 2150cm−1. Nevertheless, benzyne is more like a strained alkyne than a diradical, as seen from the large singlet–triplet gap and alkyne-like reactivity.[3]
The LUMO of aryne lies much lower than the LUMO of unstrained alkynes, which makes it a better energy match for the HOMO of nucleophiles. Hence, benzyne possesses electrophilic character and undergoes reactions with nucleophiles.[9] A detailed MO analysis of benzyne was presented in 1968.[10]
Generation of arynes
Due to their extreme reactivity, arynes must be generated in situ. Typical of other reactive intermediates, benzyne must be trapped, otherwise it dimerises to biphenylene.
Such reactions require strong base and high temperatures. 1,2-Disubstituted arenes serve as precursors to benzynes under milder conditions. Benzyne is generated by the dehalogenation of 1-bromo-2-fluorobenzene by magnesium.[11]Anthranilic acid can be converted to 2-diazoniobenzene-1-carboxylate by diazotization and neutralization. Although explosive,[12] this zwitterionic species is a convenient and inexpensive precursor to benzyne.[13]
Another method, with wide applicability, is based on trimethylsilylaryl triflates.[14][15] Fluoride displacement of the trimethylsilyl group induces elimination of triflate and release of benzyne:
Even at low temperatures arynes are extremely reactive. Their reactivity can be classified in three main classes: (1) nucleophilic additions, (2) pericyclic reactions, and (3) bond-insertion.
Nucleophilic additions to arynes
Upon treatment with basic nucleophiles, aryl halides deprotonate alpha to the leaving group, resulting in dehydrohalogenation. Isotope exchange studies indicate that for aryl fluorides and, sometimes, aryl chlorides, the elimination event proceeds in two steps, deprotonation, followed by expulsion of the nucleophile. Thus, the process is formally analogous to the E1cb mechanism of aliphatic compounds. Aryl bromides and iodides, on the other hand, generally appear to undergo elimination by a concerted syn-coplanar E2 mechanism.[18][19] The resulting benzyne forms addition products, usually by nucleophilic addition and protonation. Generation of the benzyne intermediate is the slow step in the reaction.[20]
"Aryne coupling" reactions allow for generation of biphenyl compounds which are valuable in pharmaceutical industry, agriculture and as ligands in many metal-catalyzed transformations.[21]
The metal–arene product can also add to another aryne, leading to chain-growth polymerization. Using copper(I) cyanide as the initiator to add to the first aryne yielded polymers containing up to about 100 arene units.[22]
When leaving group (LG) and substituent (Y) are mutually ortho or para, only one benzyne intermediate is possible. However, when LG is meta to Y, then regiochemical outcomes (A and B) are possible. If Y is electron withdrawing, then HB is more acidic than HA resulting in regioisomer B being generated. Analogously, if Y is electron donating, regioisomer A is generated, since now HA is the more acidic proton.
There are two possible regioisomers of benzyne with substituent (Y): triple bond can be positioned between C2 and C3 or between C3 and C4. Substituents ortho to the leaving group will lead to the triple bond between C2 and C3. Para Y and LG will lead to regioisomer with triple bond between C3 and C4. Meta substituent can afford both regioisomers as described above. Nucleophilic additions can occur with regioselectivity. Although classic explanations to explain regioselectivity refer to carbanion stability following attack by the nucleophile,[20] this explanation has been replaced by the aryne distortion model by Houk and Garg.[23][24][25] In this model, substituents cause geometric distortion of the ground state structure of the aryne, leading to regioselective reactions, consistent with reactions proceeding through early transition states.
Pericyclic reactions of arynes
Benzyne undergoes rapid dimerization to form biphenylene. Some routes to benzyne lead to especially rapid and high yield of this subsequent reaction.[13][17]Trimerization gives triphenylene.[26]
Benzynes can undergo [4+2] cyclization reactions. When generated in the presence of anthracene, trypticene results.[11] In this method, the concerted mechanism of the Diels-Alder reaction between benzyne and furan is shown below. Other benzyne [4+2] cycloadditions are thought to proceed via a stepwise mechanism.
[4+2] cycloadditions of arynes have been commonly applied to natural product total synthesis. The main limitation of such approach, however, is the need to use constrained dienes, such as furan and cyclopentadiene.[14] In 2009 Buszek and co-workers synthesized herbindole A using aryne [4+2]-cycloaddition.[29] 6,7-indolyne undergoes [4+2] cycloaddition with cyclopentadiene to afford complex tetracyclic product.
Benzynes undergo [2+2] cycloaddition with a wide range of alkenes. Due to electrophilic nature of benzyne, alkenes bearing electron-donating substituents work best for this reaction.[30]
Due to significant byproduct formation, aryne [2+2] chemistry is rarely utilized in natural product total synthesis.[14] Nevertheless, several examples do exist. In 1982, Stevens and co-workers reported a synthesis of taxodione that utilized [2+2] cycloaddition between an aryne and a ketene acetal.[31]
Mori and co-workers performed a palladium-catalyzed [2+2+2]-cocyclization of aryne and diyne in their total synthesis of taiwanins C.[32]
Bond-insertion reactions of arynes
The first example of aryne σ-bond insertion reaction is the synthesis of melleine in 1973.[33]
Other dehydrobenzenes
If benzyne is 1,2-didehydrobenzene, two further isomers are possible: 1,3-didehydrobenzene and 1,4-didehydrobenzene.[3] Their energies in silico are, respectively, 106, 122, and 138 kcal/mol (444, 510 and 577 kJ/mol).[34] The 1,2- and 1,3- isomers have singlet ground states, whereas for 1,4-didehydrobenzene the gap is smaller.
The interconversion of the 1,2-, 1,3- and 1,4-didehydrobenzenes has been studied.[34][35] A 1,2- to 1,3-didehydrobenzene conversion has been postulated to occur in the pyrolysis (900°C) of the phenyl substituted aryne precursors[34] as shown below. Extremely high temperatures are required for benzyne interconversion.
1,4-Didehydroarenes
In classical 1,4-didehydrobenzene experiments, heating to 300°C, [1,6-D2]-A readily equilibrates with [3,2-D2]-B, but does not equilibrate with C or D. The simultaneous migration of deuterium atoms to form B, and the fact that none of C or D is formed can only be explained by a presence of a cyclic and symmetrical intermediate–1,4-didehydrobenzene.[36]
Two states were proposed for 1,4-didehydrobenzene: singlet and triplet, with the singlet state lower in energy.[37][38] Triplet state represents two noninteracting radical centers, and hence should abstract hydrogens at the same rate as phenyl radical. However, singlet state is more stabilized than the triplet, and therefore some of the stabilizing energy will be lost in order to form the transition state for hydrogen cleavage, leading to slower hydrogen abstraction. Chen proposed the use of 1,4-didehydrobenzene analogues that have large singlet-triplet energy gaps to enhance selectivity of enediyne drug candidates.[39]
History
The first evidence for arynes came from the work of Stoermer and Kahlert. In 1902 they observed that upon treatment of 3-bromobenzofuran with base in ethanol 2-ethoxybenzofuran is formed. Based on this observation they postulated an aryne intermediate.[40]
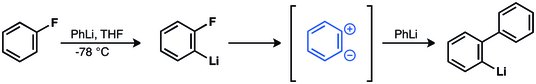
First indication of benzyne.
Wittig et al. invoked zwitterionic intermediate in the reaction of fluorobenzene and phenyllithium to give biphenyl.[41][42][43] This hypothesis was later confirmed.[44][45][46][47][48]
In 1953 14C labeling experiments provided strong support for the intermediacy of benzyne.[44]John D. Roberts et al. showed that the reaction of chlorobenzene-1-14C and potassium amide gave equal amounts of aniline with 14C incorporation at C-1 and C-2.
C labeling experiment shows equal distribution of products.
Wittig and Pohmer found that benzyne participate in [4+2] cycloaddition reactions.[49]
Capture of benzyne as dienophile in Diels-Alder reaction.
Additional evidence for the existence of benzyne came from spectroscopic studies.[3] Benzyne has been observed in a "molecular container".[50]
In 2015, a single aryne molecule was imaged by STM.[51]
1,3-Didehydroarenes was first demonstrated in the 1990s when it was generated from 1,3-disubstituted benzene derivatives, such as the peroxy ester 1,3-C6H4(O2C(O)CH3)2.[3]
Breakthroughs on 1,4-didehydrobenzene came in the 1960s, followed from studies on the Bergman cyclization.[36] This theme became topical with the discovery of enediyne "cytostatics", such as calicheamicin, which generates a 1,4-didehydrobenzene.[52]
Examples of benzynes in total synthesis
A variety of natural products have been prepared using arynes as intermediates.[14] Nucleophilic additions to arynes have been widely used in natural product total synthesis. Indeed, nucleophilic additions of arynes are some of the oldest known applications of aryne chemistry.[14] Nucleophilic addition to aryne was used in the attempted synthesis of cryptaustoline (1) and cryptowoline (2).[53]
The synthesis of the tetracyclic meroterpenoid (+)-liphagal involved an aryne intermediate.[54] Their approach employed an aryne cyclization to close the final ring of the natural product.[14]
Multicomponent reactions of arynes are powerful transformations that allow for rapid formation of 1,2-disubstituted arenes. Despite their potential utility, examples of multicomponent aryne reactions in natural product synthesis are scarce.[14] A four-component aryne coupling reaction was employed in the synthesis of dehydroaltenuene B.[55]
↑Radziszewski, J. G.; Hess, B. A. Jr.; Zahradnik, R. (1992). "Infrared Spectrum of o-Benzyne: Experiment and Theory". J. Am. Chem. Soc. 114 (1): 52. Bibcode:1992JAChS.114...52R. doi:10.1021/ja00027a007.
↑Gilchrist, T. L. Supplement C: The Chemistry of Triple Bonded Functional Groups, Part 1. Patai, S.; Rappaport, Z. Eds., John Wiley & Sons, New York, 1983
↑Hoffmann, R.; Imamura, A.; Hehre, W. J. (1968). "Benzynes, dehydroconjugated molecules, and the interaction of orbitals separated by a number of intervening sigma bonds". J. Am. Chem. Soc. 90 (6): 1499. Bibcode:1968JAChS..90.1499H. doi:10.1021/ja01008a018.
1234567Tadross, P. M.; Stoltz, B. M. (2012). "A Comprehensive History of Arynes in Natural Product Total Synthesis". Chem. Rev. 112 (6): 3550–3577. doi:10.1021/cr200478h. PMID22443517.
↑Panar, Manuel (1961). The Elimination-Addition Mechanism of Nucleophilic Aromatic Substitution. Pasadena, CA: California Institute of Technology (Ph.D. Thesis). pp.4–5.
↑"Use of 1,2,4,5-Tetrabromobenzene as a 1,4-Nenzadiyne Equivalent: Anti- and Syn-1,4,5,8-tetrahydroanthracene 1,4:5,8-diepoxides". Organic Syntheses. 75: 201. 1998. doi:10.15227/orgsyn.075.0201.
↑Polishchuk, A. L.; Bartlett, K. L.; Friedman, L. A.; Jones, M. Jr (2004). "A p-Benzyne to m-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Completion of the Benzyne Cascade". J. Phys. Org. Chem. 17 (9): 798–806. doi:10.1002/poc.797.
12Richard R. Jones; Robert G. Bergman (1972). "p-Benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure". J. Am. Chem. Soc.94 (2): 660–661. Bibcode:1972JAChS..94..660J. doi:10.1021/ja00757a071.
↑Clauberg, H.; Minsek, D. W.; Chen, P. (1992). "Mass and photoelectron spectroscopy of C3H2. .DELTA.Hf of singlet carbenes deviate from additivity by their singlet-triplet gaps". J. Am. Chem. Soc. 114 (1): 99. Bibcode:1992JAChS.114...99C. doi:10.1021/ja00027a014.
↑Blush, J. A.; Clauberg, H.; Kohn, D. W.; Minsek, D. W.; Zhang, X.; Chen, P. (1992). "Photoionization mass and photoelectron spectroscopy of radicals, carbenes, and biradicals". Acc. Chem. Res. 25 (9): 385. doi:10.1021/ar00021a001.
↑Wittig, G.; Pieper, G.; Fuhrmann, G. (1940). "Über die Bildung von Diphenyl aus Fluorbenzol und Phenyl-lithium (IV. Mitteil. über Austauschreaktionen mit Phenyl-lithium)". Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 73 (11): 1193–1197. doi:10.1002/cber.19400731113.
12Roberts, John D. (1953). "Rearrangement in the Reaction of Chlorobenzene-1-C14With Potassium Amide1". Journal of the American Chemical Society. 75 (13): 3290–3291. Bibcode:1953JAChS..75.3290R. doi:10.1021/ja01109a523.
↑Roberts, John D. (1956). "Orientation in Aminations of Substituted Halobenzenes 1". Journal of the American Chemical Society. 78 (3): 611–614. Bibcode:1956JAChS..78..611R. doi:10.1021/ja01584a025.
↑Soorukram, D.; Qu, T.; Barrett, A. G. M. (2008). "Four-Component Benzyne Coupling Reactions: A Concise Total Synthesis of Dehydroaltenuene B". Org. Lett. 10 (17): 3833–3835. doi:10.1021/ol8015435. PMID18672878.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.