
Achondrogenesis, type 2 is an uncommon skeletal dysplasia that is autosomal dominant and occurs at a frequency of approximately 0.2 per 100,000 births. Also known by the name Langer-Saldino achondrogenesis, it is one of the fatal short-limbed dwarfisms linked to structural mutations in type II collagen.
Barakat syndrome is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It is an autosomal dominant condition with incomplete penetrance and variable expressivity that was first described by Amin J. Barakat et al. in 1977.
Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.

Filamin B, beta (FLNB), also known as Filamin B, beta , is a cytoplasmic protein which in humans is encoded by the FLNB gene.
Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.

Fibrochondrogenesis is a rare autosomal recessive form of osteochondrodysplasia, causing abnormal fibrous development of cartilage and related tissues.
X-linked recessive chondrodysplasia punctata is a type of chondrodysplasia punctata that can involve the skin, hair, and cause short stature with skeletal abnormalities, cataracts, and deafness.
A Finnish heritage disease is any genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Northern Sweden (Meänmaa) and Northwest Russia. There are 36 rare diseases regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis, a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones. The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.
Bruck syndrome is characterized as the combination of arthrogryposis multiplex congenita and osteogenesis imperfecta. Both diseases are uncommon, but concurrence is extremely rare which makes Bruck syndrome very difficult to research. Bruck syndrome is thought to be an atypical variant of osteogenesis imperfecta most resembling type III, if not its own disease. Multiple gene mutations associated with osteogenesis imperfecta are not seen in Bruck syndrome. Many affected individuals are within the same family, and pedigree data supports that the disease is acquired through autosomal recessive inheritance. Bruck syndrome has features of congenital contractures, bone fragility, recurring bone fractures, flexion joint and limb deformities, pterygia, short body height, and progressive kyphoscoliosis. Individuals encounter restricted mobility and pulmonary function. A reduction in bone mineral content and larger hydroxyapatite crystals are also detectable Joint contractures are primarily bilateral and symmetrical, and most prone to ankles. Bruck syndrome has no effect on intelligence, vision, or hearing.
Acro–dermato–ungual–lacrimal–tooth syndrome is a rare genetic disease. It is an autosomal dominant form of ectodermal dysplasia, a group of disorders that affects the hair, teeth, nails, sweat glands, and extremities. The syndrome arises from a mutation in the TP63 gene. This disease was previously thought to be a form of ectrodactyly–ectodermal dysplasia–cleft syndrome (EEC), but was classified as a different disease in 1993 by Propping and Zerres.
Hecht Scott syndrome is a rare genetic disease that causes congenital limb formation. The main characterisation is the aplasia or hypoplasia of bones of the limb. It is currently presenting in less than 1 in 1,000,000 newborns. It has been known to be more commonly present in males. It was first diagnosed in 2005 by Courtens et al. who recognised the malformations with his present case and four others that were similarly described in literature.
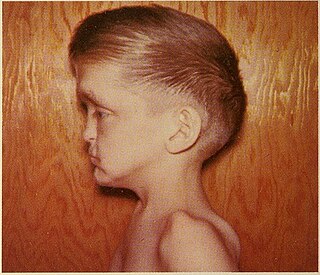
Oto-palato-digital syndrome is the generalised term for two conditions, oto-palato-digital syndrome type I (OPD1) and oto-palato-digital syndrome type II (OPD2), that are both X-linked recessive genetic disorders with overlapping phenotypes. The most severe phenotypes of each syndrome occur only in males, with females generally having attenuated forms of the condition, although this does not apply to all individual cases. Some writers conceptualise oto-palato-digital syndrome as a spectrum disorder including two similarly-presenting genetic syndromes, frontometaphyseal dysplasia and Melnick-Needles syndrome.
Meacham syndrome is a rare genetic disorder which is characterized by lung, diaphragmatic and genitourinary anomalies.
Spondyloepimetaphyseal dysplasia-short limb-abnormal calcification syndrome is a rare genetic disorder which is characterized by osseous anomalies resulting in short stature and other afflictions.
Czech dysplasia metatarsal type is a rare type of Czech dysplasia which is characterized primarily by bone anomalies.
Holoprosencephaly-ectrodactyly-cleft lip/palate syndrome, also simply known as Hartsfield syndrome, is a rare genetic disorder characterized by the presence of variable holoprosencephaly, ectrodactyly, cleft lip and palate, alongside generalized ectodermal abnormalities. Additional findings include endocrine anomalies and developmental delays.
Schneckenbecken dysplasia is a rare pre-natally fatal hereditary autosomal recessive condition which affects the bones and pre-natal growth.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
