Distinctive feature, known as a derived trait, that is unique to a given taxon
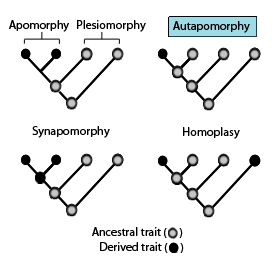
Phylogenies showing the terminology used to describe different patterns of ancestral and derived character or trait states (such as the evolution of a flatfish).
In phylogenetics, an autapomorphy is a distinctive feature, known as a derived trait, that is unique to a given taxon. That is, it is found only in one taxon, but not found in any others or outgrouptaxa, not even those most closely related to the focal taxon (which may be a species, family or in general any clade).[2] It can therefore be considered as an apomorphy in relation to a single taxon.[3] The word autapomorphy, introduced in 1950 by German entomologistWilli Hennig, is derived from the Greek words αὐτός, autos "self"; ἀπό, apo "away from"; and μορφή, morphḗ = "shape".[4]
Background
Because autapomorphies are only present in a single taxon, they do not convey information about relationship. Therefore, autapomorphies are not useful to infer phylogenetic relationships. However, autapomorphy, like synapomorphy and plesiomorphy is a relative concept depending on the taxon in question. An autapomorphy at a given level may well be a synapomorphy at a less-inclusive level.[5] An example of an autapomorphy can be described in modern snakes. Snakes have lost the two pairs of legs that characterize all of Tetrapoda, and the closest taxa to Ophidia – as well as their common ancestors – all have two pairs of legs. Therefore, the Ophidia taxon presents an autapomorphy with respect to its absence of legs.[3]
The autapomorphic species concept is one of many methods that scientists might use to define and distinguish species from one another. This definition assigns species on the basis of amount of divergence associated with reproductive incompatibility, which is measured essentially by number of autapomorphies.[6] This grouping method is often referred to as the "monophyletic species concept" or the "phylospecies" concept and was popularized by D.E. Rosen in 1979. Within this definition, a species is seen as "the least inclusive monophyletic group definable by at least one autapomorphy".[7] While this model of speciation is useful in that it avoids non-monophyletic groupings, it has its criticisms as well. N.I. Platnick, for example, believes the autapomorphic species concept to be inadequate because it allows for the possibility of reproductive isolation and speciation while revoking the "species" status of the mother population. In other words, if a peripheral population breaks away and becomes reproductively isolated, it would conceivably need to develop at least one autapomorphy to be recognized as a different species. If this can happen without the larger mother population also developing a new autapomorphy, then the mother population cannot remain a species under the autapomorphic species concept: it would no longer have any apomorphies not also shared by the daughter species.[8]
Phylogenetic similarities: These phylogenetic terms are used to describe different patterns of ancestral and derived character ortraitstates as stated in the above diagram in association with synapomorphies.[1]
Homoplasy in biological systematics is when a trait has been gained or lost independently in separate lineages during evolution. This convergent evolution leads to species independently sharing a trait that is different from the trait inferred to have been present in their common ancestor.[9][10][11]
Reverse Homoplasy – trait present in an ancestor but not in direct descendants that reappears in later descendants.[13]
Apomorphy – a derived trait. Apomorphy shared by two or more taxa and inherited from a common ancestor is synapomorphy. Apomorphy unique to a given taxon is autapomorphy.[14][15][16][17]
Synapomorphy/Homology – a derived trait that is found in some or all terminal groups of a clade, and inherited from a common ancestor, for which it was an autapomorphy (i.e., not present in its immediate ancestor).
Underlying synapomorphy – a synapomorphy that has been lost again in many members of the clade. If lost in all but one, it can be hard to distinguish from an autapomorphy.
Autapomorphy – a distinctive derived trait that is unique to a given taxon or group.[3]
Symplesiomorphy – an ancestral trait shared by two or more taxa.
Plesiomorphy – a symplesiomorphy discussed in reference to a more derived state.
Pseudoplesiomorphy – is a trait that cannot be identified as neither a plesiomorphy nor an apomorphy that is a reversal.[18]
Reversal – is a loss of derived trait present in ancestor and the reestablishment of a plesiomorphic trait.
Convergence – independent evolution of a similar trait in two or more taxa.
↑ Futuyma DJ (1998). Evolutionary Biology (3rded.). Sinauer Associates, Inc. p.95.
1 2 3 Appel RD, Feytmans E (2009). "Chapter 3: Introduction of Phylogenetics and its Molecular Aspects". Bioinformatics: a Swiss Perspective (1sted.). World Scientific Publishing Company.
↑ Archie JW (September 1989). "Homoplasy Excess Ratios: New Indices for Measuring Levels of Homoplasy in Phylogenetic Systematics and a Critique of the Consistency Index". Systematic Zoology. 38 (3): 253–269. doi:10.2307/2992286. JSTOR2992286.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.