
An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine. Enamines are versatile intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
The Robinson annulation is a chemical reaction used in organic chemistry for ring formation. It was discovered by Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds. The method uses a ketone and a methyl vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring by a Michael addition followed by an aldol condensation. This procedure is one of the key methods to form fused ring systems.

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being collectively awarded one half of the 2001 Nobel Prize in Chemistry.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
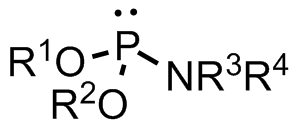
A phosphoramidite (RO)2PNR2 is a monoamide of a phosphite diester. The key feature of phosphoramidites is their markedly high reactivity towards nucleophiles catalyzed by weak acids e.c., triethylammonium chloride or 1H-tetrazole. In these reactions, the incoming nucleophile replaces the NR2 moiety.
Within the area of organocatalysis, (thio)urea organocatalysis describes the use of ureas and thioureas to accelerate and stereochemically alter organic transformations. The effects arise through hydrogen-bonding interactions between the substrate and the (thio)urea. Unlike classical catalysts, these organocatalysts interact by non-covalent interactions, especially hydrogen bonding. The scope of these small-molecule H-bond donors termed (thio)urea organocatalysis covers both non-stereoselective and stereoselective reactions.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. The field is relatively undeveloped compared to research in Lewis acid catalysis.
In organic chemistry, the Keck asymmetric allylation is a chemical reaction that involves the nucleophilic addition of an allyl group to an aldehyde. The catalyst is a chiral complex that contains titanium as a Lewis acid. The chirality of the catalyst induces a stereoselective addition, so the secondary alcohol of the product has a predictable absolute stereochemistry based on the choice of catalyst. This name reaction is named for Gary Keck.

Ugi’s amine is an organometallic compound with the formula (C5H5)Fe(C5H4CH N 2. It is named for the chemist who first reported its synthesis in 1970, Ivar Ugi. It is a ferrocene derivative. Ugi’s amine is a precursor to ligands, most notably, the Josiphos ligands, which have been used in asymmetric catalysis
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.


