
A neurotransmitter is a signaling molecule secreted by a neuron to affect another cell across a synapse. The cell receiving the signal, or target cell, may be another neuron, but could also be a gland or muscle cell.

Acetylcholine (ACh) is an organic compound that functions in the brain and body of many types of animals as a neurotransmitter. Its name is derived from its chemical structure: it is an ester of acetic acid and choline. Parts in the body that use or are affected by acetylcholine are referred to as cholinergic.
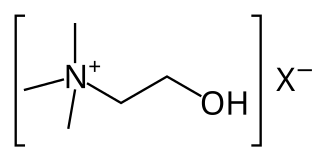
Cholinergic agents are compounds which mimic the action of acetylcholine and/or butyrylcholine. In general, the word "choline" describes the various quaternary ammonium salts containing the N,N,N-trimethylethanolammonium cation. Found in most animal tissues, choline is a primary component of the neurotransmitter acetylcholine and functions with inositol as a basic constituent of lecithin. Choline also prevents fat deposits in the liver and facilitates the movement of fats into cells.

The suprachiasmatic nucleus or nuclei (SCN) is a small region of the brain in the hypothalamus, situated directly above the optic chiasm. The SCN is the principal circadian pacemaker in mammals, responsible for generating circadian rhythms. Reception of light inputs from photosensitive retinal ganglion cells allow the SCN to coordinate the subordinate cellular clocks of the body and entrain to the environment. The neuronal and hormonal activities it generates regulate many different body functions in an approximately 24-hour cycle.

Choline acetyltransferase is a transferase enzyme responsible for the synthesis of the neurotransmitter acetylcholine. ChAT catalyzes the transfer of an acetyl group from the coenzyme acetyl-CoA to choline, yielding acetylcholine (ACh). ChAT is found in high concentration in cholinergic neurons, both in the central nervous system (CNS) and peripheral nervous system (PNS). As with most nerve terminal proteins, ChAT is produced in the body of the neuron and is transported to the nerve terminal, where its concentration is highest. Presence of ChAT in a nerve cell classifies this cell as a "cholinergic" neuron. In humans, the choline acetyltransferase enzyme is encoded by the CHAT gene.

The locus coeruleus (LC), also spelled locus caeruleus or locus ceruleus, is a nucleus in the pons of the brainstem involved with physiological responses to stress and panic. It is a part of the reticular activating system.

Coluracetam is a purported nootropic agent of the racetam family. It is contains a chemical group that is a bioisostere of the 9-amino-tetrahydroacridine family. It was initially developed and tested by the Mitsubishi Tanabe Pharma Corporation for Alzheimer's disease. After the drug failed to reach endpoints in its clinical trials it was in-licensed by BrainCells Inc for investigations into major depressive disorder (MDD), which was preceded by being awarded a "Qualifying Therapeutic Discovery Program Grant" by the state of California. Findings from phase IIa clinical trials have suggested that it would be a potential medication for comorbid MDD with generalized anxiety disorder (GAD). BrainCells Inc is currently out-licensing the drug for this purpose. It may also have potential use in prevention and treatment of ischemic retinopathy and retinal and optic nerve injury.
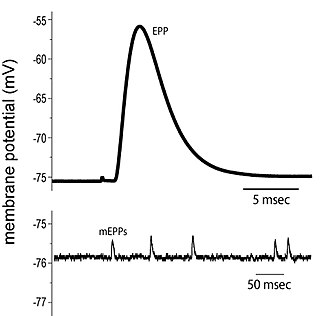
End plate potentials (EPPs) are the voltages which cause depolarization of skeletal muscle fibers caused by neurotransmitters binding to the postsynaptic membrane in the neuromuscular junction. They are called "end plates" because the postsynaptic terminals of muscle fibers have a large, saucer-like appearance. When an action potential reaches the axon terminal of a motor neuron, vesicles carrying neurotransmitters are exocytosed and the contents are released into the neuromuscular junction. These neurotransmitters bind to receptors on the postsynaptic membrane and lead to its depolarization. In the absence of an action potential, acetylcholine vesicles spontaneously leak into the neuromuscular junction and cause very small depolarizations in the postsynaptic membrane. This small response (~0.4mV) is called a miniature end plate potential (MEPP) and is generated by one acetylcholine-containing vesicle. It represents the smallest possible depolarization which can be induced in a muscle.

The ventrolateral preoptic nucleus (VLPO), also known as the intermediate nucleus of the preoptic area (IPA), is a small cluster of neurons situated in the anterior hypothalamus, sitting just above and to the side of the optic chiasm in the brain of humans and other animals. The brain's sleep-promoting nuclei, together with the ascending arousal system which includes components in the brainstem, hypothalamus and basal forebrain, are the interconnected neural systems which control states of arousal, sleep, and transitions between these two states. The VLPO is active during sleep, particularly during non-rapid eye movement sleep, and releases inhibitory neurotransmitters, mainly GABA and galanin, which inhibit neurons of the ascending arousal system that are involved in wakefulness and arousal. The VLPO is in turn innervated by neurons from several components of the ascending arousal system. The VLPO is activated by the endogenous sleep-promoting substances adenosine and prostaglandin D2. The VLPO is inhibited during wakefulness by the arousal-inducing neurotransmitters norepinephrine and acetylcholine. The role of the VLPO in sleep and wakefulness, and its association with sleep disorders – particularly insomnia and narcolepsy – is a growing area of neuroscience research.

Part of the human brain, the basal forebrain structures are located in the forebrain to the front of and below the striatum. They include the ventral basal ganglia, nucleus basalis, diagonal band of Broca, substantia innominata, and the medial septal nucleus. These structures are important in the production of acetylcholine, which is then distributed widely throughout the brain. The basal forebrain is considered to be the major cholinergic output of the central nervous system (CNS) centred on the output of the nucleus basalis. The presence of non-cholinergic neurons projecting to the cortex have been found to act with the cholinergic neurons to dynamically modulate activity in the cortex.
Neuromodulation is the physiological process by which a given neuron uses one or more chemicals to regulate diverse populations of neurons. Neuromodulators typically bind to metabotropic, G-protein coupled receptors (GPCRs) to initiate a second messenger signaling cascade that induces a broad, long-lasting signal. This modulation can last for hundreds of milliseconds to several minutes. Some of the effects of neuromodulators include: altering intrinsic firing activity, increasing or decreasing voltage-dependent currents, altering synaptic efficacy, increasing bursting activity and reconfigurating synaptic connectivity.
The biochemistry of Alzheimer's disease, the most common cause of dementia, is not yet very well understood. Alzheimer's disease (AD) has been identified as a proteopathy: a protein misfolding disease due to the accumulation of abnormally folded amyloid beta (Aβ) protein in the brain. Amyloid beta is a short peptide that is an abnormal proteolytic byproduct of the transmembrane protein amyloid-beta precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of proteolytic complex involved in APP processing and degradation.
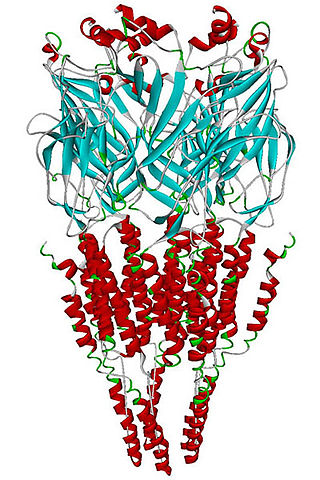
The alpha-7 nicotinic receptor, also known as the α7 receptor, is a type of nicotinic acetylcholine receptor implicated in long-term memory, consisting entirely of α7 subunits. As with other nicotinic acetylcholine receptors, functional α7 receptors are pentameric [i.e., (α7)5 stoichiometry].

Alzheimer's disease (AD) is a neurodegenerative disease that usually starts slowly and progressively worsens, and is the cause of 60–70% of cases of dementia. The most common early symptom is difficulty in remembering recent events. As the disease advances, symptoms can include problems with language, disorientation, mood swings, loss of motivation, self-neglect, and behavioral issues. As a person's condition declines, they often withdraw from family and society. Gradually, bodily functions are lost, ultimately leading to death. Although the speed of progression can vary, the typical life expectancy following diagnosis is three to nine years.
Sleep onset is the transition from wakefulness into sleep. Sleep onset usually transmits into non-rapid eye movement sleep but under certain circumstances it is possible to transit from wakefulness directly into rapid eye movement sleep.

Clinical neurochemistry is the field of neurological biochemistry which relates biochemical phenomena to clinical symptomatic manifestations in humans. While neurochemistry is mostly associated with the effects of neurotransmitters and similarly functioning chemicals on neurons themselves, clinical neurochemistry relates these phenomena to system-wide symptoms. Clinical neurochemistry is related to neurogenesis, neuromodulation, neuroplasticity, neuroendocrinology, and neuroimmunology in the context of associating neurological findings at both lower and higher level organismal functions.

In the human brain, the nucleus basalis, also known as the nucleus basalis of Meynert or nucleus basalis magnocellularis, is a group of neurons located mainly in the substantia innominata of the basal forebrain. Most neurons of the nucleus basalis are rich in the neurotransmitter acetylcholine, and they have widespread projections to the neocortex and other brain structures.

The neuroscience of sleep is the study of the neuroscientific and physiological basis of the nature of sleep and its functions. Traditionally, sleep has been studied as part of psychology and medicine. The study of sleep from a neuroscience perspective grew to prominence with advances in technology and the proliferation of neuroscience research from the second half of the twentieth century.

A neuronal lineage marker is an endogenous tag that is expressed in different cells along neurogenesis and differentiated cells such as neurons. It allows detection and identification of cells by using different techniques. A neuronal lineage marker can be either DNA, mRNA or RNA expressed in a cell of interest. It can also be a protein tag, as a partial protein, a protein or an epitope that discriminates between different cell types or different states of a common cell. An ideal marker is specific to a given cell type in normal conditions and/or during injury. Cell markers are very valuable tools for examining the function of cells in normal conditions as well as during disease. The discovery of various proteins specific to certain cells led to the production of cell-type-specific antibodies that have been used to identify cells.
Robert Y. Moore is an American neurologist with interests in disorders of biological rhythms, movement disorders, and behavioral neurology. He is credited with discovering the function of the suprachiasmatic nucleus (SCN) as the circadian clock, as well as, describing its organization. He is also credited with establishing the role of the mammalian retinohypothalamic tract (RHT) as a photic entrainment pathway. Moore cin 2017 serves as a professor of neurology, with a secondary in psychiatry and neuroscience at the University of Pittsburgh, and as co-director of the National Parkinson Foundation Center of Excellence at the University of Pittsburgh.
