
Structural biology, as defined by the Journal of Structural Biology, deals with structural analysis of living material at every level of organization. Early structural biologists throughout the 19th and early 20th centuries were primarily only able to study structures to the limit of the naked eye's visual acuity and through magnifying glasses and light microscopes.

Wolfram Research, Inc. is an American multinational company that creates computational technology. Wolfram's flagship product is the technical computing program Wolfram Mathematica, first released on June 23, 1988. Other products include WolframAlpha, Wolfram SystemModeler, Wolfram Workbench, gridMathematica, Wolfram Finance Platform, webMathematica, the Wolfram Cloud, and the Wolfram Programming Lab. Wolfram Research founder Stephen Wolfram is the CEO. The company is headquartered in Champaign, Illinois, United States.
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.
Bioconductor is a free, open source and open development software project for the analysis and comprehension of genomic data generated by wet lab experiments in molecular biology.

IGOR Pro is a scientific data analysis software, numerical computing environment and programming language that runs on Windows or Mac operating systems. It is developed by WaveMetrics Inc., and was originally aimed at time series analysis, but has since then evolved and covers other applications such as curve fitting and image processing. It comes with a fully functional programming language and compiler, but many functions are also accessible through menus. IGOR Pro is primarily known for its graphics capabilities, and like Origin and other similar programs, is often used to generate plots for scientific and other publications. Other features include the possibility of extending the built-in functions with external operations (XOP) allowing data acquisition, manipulation and analysis features, communication with external devices and in principle any other task that can be programmed in C or C++.
The Earth System Modeling Framework (ESMF) is open-source software for building climate, numerical weather prediction, data assimilation, and other Earth science software applications. These applications are computationally demanding and usually run on supercomputers. The ESMF is considered a technical layer, integrated into a sophisticated common modeling infrastructure for interoperability. Other aspects of interoperability and shared infrastructure include: common experimental protocols, common analytic methods, common documentation standards for data and data provenance, shared workflow, and shared model components.

PQS is a general purpose quantum chemistry program. Its roots go back to the first ab initio gradient program developed in Professor Peter Pulay's group but now it is developed and distributed commercially by Parallel Quantum Solutions. There is a reduction in cost for academic users and a site license. Its strong points are geometry optimization, NMR chemical shift calculations, and large MP2 calculations, and high parallel efficiency on computing clusters. It includes many other capabilities including Density functional theory, the semiempirical methods, MINDO/3, MNDO, AM1 and PM3, Molecular mechanics using the SYBYL 5.0 Force Field, the quantum mechanics/molecular mechanics mixed method using the ONIOM method, natural bond orbital (NBO) analysis and COSMO solvation models. Recently, a highly efficient parallel CCSD(T) code for closed shell systems has been developed. This code includes many other post Hartree–Fock methods: MP2, MP3, MP4, CISD, CEPA, QCISD and so on.
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH, and by Ad Bax, Marius Clore, Angela Gronenborn at the NIH, and Gerhard Wagner at Harvard University, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
X-PLOR is a computer software package for computational structural biology originally developed by Axel T. Brunger at Yale University. It was first published in 1987 as an offshoot of CHARMM - a similar program that ran on supercomputers made by Cray Inc. It is used in the fields of X-ray crystallography and nuclear magnetic resonance spectroscopy of proteins (NMR) analysis.
The Collaborative Computational Project Number 4 in Protein Crystallography (CCP4) was set up in 1979 in the United Kingdom to support collaboration between researchers working in software development and assemble a comprehensive collection of software for structural biology. The CCP4 core team is located at the Research Complex at Harwell (RCaH) at Rutherford Appleton Laboratory (RAL) in Didcot, near Oxford, UK.
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
In software engineering, a software development process or software development life cycle (SDLC) is a process of planning and managing software development. It typically involves dividing software development work into smaller, parallel, or sequential steps or sub-processes to improve design and/or product management. The methodology may include the pre-definition of specific deliverables and artifacts that are created and completed by a project team to develop or maintain an application.
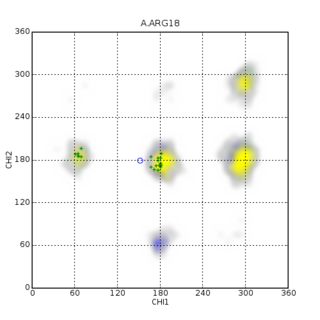
In biomolecular structure, CING stands for the Common Interface for NMR structure Generation and is known for structure and NMR data validation.

Antony John Williams is a British chemist and expert in the fields of both nuclear magnetic resonance (NMR) spectroscopy and cheminformatics at the United States Environmental Protection Agency. He is the founder of the ChemSpider website that was purchased by the Royal Society of Chemistry in May 2009. He is a science blogger and an author.
The Re-referenced Protein Chemical shift Database (RefDB) is an NMR spectroscopy database of carefully corrected or re-referenced chemical shifts, derived from the BioMagResBank (BMRB). The database was assembled by using a structure-based chemical shift calculation program to calculate expected protein (1)H, (13)C and (15)N chemical shifts from X-ray or NMR coordinate data of previously assigned proteins reported in the BMRB. The comparison is automatically performed by a program called SHIFTCOR. The RefDB database currently provides reference-corrected chemical shift data on more than 2000 assigned peptides and proteins. Data from the database indicates that nearly 25% of BMRB entries with (13)C protein assignments and 27% of BMRB entries with (15)N protein assignments require significant chemical shift reference readjustments. Additionally, nearly 40% of protein entries deposited in the BioMagResBank appear to have at least one assignment error. Users may download, search or browse the database through a number of methods available through the RefDB website. RefDB provides a standard chemical shift resource for biomolecular NMR spectroscopists, wishing to derive or compute chemical shift trends in peptides and proteins.

WeNMR is a worldwide e-Infrastructure for NMR spectroscopy and structural biology. It is the largest virtual Organization in the life sciences and is supported by EGI.
A Benchtop nuclear magnetic resonance spectrometer refers to a Fourier transform nuclear magnetic resonance (FT-NMR) spectrometer that is significantly more compact and portable than the conventional equivalents, such that it is portable and can reside on a laboratory benchtop. This convenience comes from using permanent magnets, which have a lower magnetic field and decreased sensitivity compared to the much larger and more expensive cryogen cooled superconducting NMR magnets. Instead of requiring dedicated infrastructure, rooms and extensive installations these benchtop instruments can be placed directly on the bench in a lab and moved as necessary. These spectrometers offer improved workflow, even for novice users, as they are simpler and easy to use. They differ from relaxometers in that they can be used to measure high resolution NMR spectra and are not limited to the determination of relaxation or diffusion parameters.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
