Complete-linkage clustering is one of several methods of agglomerative hierarchical clustering. At the beginning of the process, each element is in a cluster of its own. The clusters are then sequentially combined into larger clusters until all elements end up being in the same cluster. The method is also known as farthest neighbour clustering. The result of the clustering can be visualized as a dendrogram, which shows the sequence of cluster fusion and the distance at which each fusion took place.[1][2][3]
At each step, the two clusters separated by the shortest distance are combined. The definition of 'shortest distance' is what differentiates between the different agglomerative clustering methods. In complete-linkage clustering, the link between two clusters contains all element pairs, and the distance between clusters equals the distance between those two elements (one in each cluster) that are farthest away from each other. The shortest of these links that remains at any step causes the fusion of the two clusters whose elements are involved.
Mathematically, the complete linkage function — the distance between clusters and — is described by the following expression:
where
is the distance between elements and ;
and are two sets of elements (clusters).
Algorithms
Naive scheme
The following algorithm is an agglomerative scheme that erases rows and columns in a proximity matrix as old clusters are merged into new ones. The proximity matrix D contains all distances d(i,j). The clusterings are assigned sequence numbers 0,1,......, (n−1) and L(k) is the level of the kth clustering. A cluster with sequence number m is denoted (m) and the proximity between clusters (r) and (s) is denoted d[(r),(s)].
The complete linkage clustering algorithm consists of the following steps:
Begin with the disjoint clustering having level and sequence number .
Find the most similar pair of clusters in the current clustering, say pair , according to where the maximum is over all pairs of clusters in the current clustering.
Increment the sequence number: . Merge clusters and into a single cluster to form the next clustering . Set the level of this clustering to
Update the proximity matrix, , by deleting the rows and columns corresponding to clusters and and adding a row and column corresponding to the newly formed cluster. The proximity between the new cluster, denoted , and an old cluster is defined as .
If all objects are in one cluster, stop. Else, go to step 2.
Optimally efficient scheme
The algorithm explained above is easy to understand but of complexity . In May 1976, D. Defays proposed an optimally efficient algorithm of only complexity known as CLINK (published 1977)[4] inspired by the similar algorithm SLINK for single-linkage clustering.
This section needs expansion. You can help by adding to it. (October 2011)
Let us assume that we have five elements and the following matrix of pairwise distances between them:
a
b
c
d
e
a
0
17
21
31
23
b
17
0
30
34
21
c
21
30
0
28
39
d
31
34
28
0
43
e
23
21
39
43
0
In this example, is the smallest value of , so we join elements and .
First branch length estimation
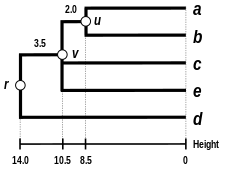
Let denote the node to which and are now connected. Setting ensures that elements and are equidistant from . This corresponds to the expectation of the ultrametricity hypothesis. The branches joining and to then have lengths (see the final dendrogram)
First distance matrix update
We then proceed to update the initial proximity matrix into a new proximity matrix (see below), reduced in size by one row and one column because of the clustering of with . Bold values in correspond to the new distances, calculated by retaining the maximum distance between each element of the first cluster and each of the remaining elements:
Italicized values in are not affected by the matrix update as they correspond to distances between elements not involved in the first cluster.
Second step
Second clustering
We now reiterate the three previous steps, starting from the new distance matrix :
(a,b)
c
d
e
(a,b)
0
30
34
23
c
30
0
28
39
d
34
28
0
43
e
23
39
43
0
Here, is the lowest value of , so we join cluster with element .
Second branch length estimation
Let denote the node to which and are now connected. Because of the ultrametricity constraint, the branches joining or to , and to , are equal and have the following total length:
We then proceed to update the matrix into a new distance matrix (see below), reduced in size by one row and one column because of the clustering of with :
Third step
Third clustering
We again reiterate the three previous steps, starting from the updated distance matrix .
((a,b),e)
c
d
((a,b),e)
0
39
43
c
39
0
28
d
43
28
0
Here, is the smallest value of , so we join elements and .
Third branch length estimation
Let denote the node to which and are now connected. The branches joining and to then have lengths (see the final dendrogram)
Third distance matrix update
There is a single entry to update:
Final step
The final matrix is:
((a,b),e)
(c,d)
((a,b),e)
0
43
(c,d)
43
0
So we join clusters and .
Let denote the (root) node to which and are now connected. The branches joining and to then have lengths:
We deduce the two remaining branch lengths:
The complete-linkage dendrogram
WPGMA Dendrogram 5S data
The dendrogram is now complete. It is ultrametric because all tips ( to ) are equidistant from :
The dendrogram is therefore rooted by , its deepest node.
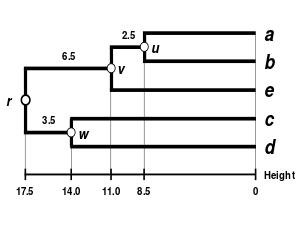
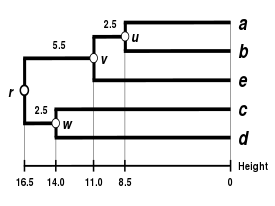
Comparison with other linkages
Alternative linkage schemes include single linkage clustering and average linkage clustering - implementing a different linkage in the naive algorithm is simply a matter of using a different formula to calculate inter-cluster distances in the initial computation of the proximity matrix and in step 4 of the above algorithm. An optimally efficient algorithm is however not available for arbitrary linkages. The formula that should be adjusted has been highlighted using bold text.
Complete linkage clustering avoids a drawback of the alternative single linkage method - the so-called chaining phenomenon, where clusters formed via single linkage clustering may be forced together due to single elements being close to each other, even though many of the elements in each cluster may be very distant to each other. Complete linkage tends to find compact clusters of approximately equal diameters.[7]
Comparison of dendrograms obtained under different clustering methods from the same distance matrix.
Kinematics is a subfield of physics and mathematics, developed in classical mechanics, that describes the motion of points, bodies (objects), and systems of bodies without considering the forces that cause them to move. Kinematics, as a field of study, is often referred to as the "geometry of motion" and is occasionally seen as a branch of both applied and pure mathematics since it can be studied without considering the mass of a body or the forces acting upon it. A kinematics problem begins by describing the geometry of the system and declaring the initial conditions of any known values of position, velocity and/or acceleration of points within the system. Then, using arguments from geometry, the position, velocity and acceleration of any unknown parts of the system can be determined. The study of how forces act on bodies falls within kinetics, not kinematics. For further details, see analytical dynamics.
The moment of inertia, otherwise known as the mass moment of inertia, angular/rotational mass, second moment of mass, or most accurately, rotational inertia, of a rigid body is a quantity that determines the torque needed for a desired angular acceleration about a rotational axis, akin to how mass determines the force needed for a desired acceleration. It depends on the body's mass distribution and the axis chosen, with larger moments requiring more torque to change the body's rate of rotation by a given amount.
In bioinformatics, neighbor joining is a bottom-up (agglomerative) clustering method for the creation of phylogenetic trees, created by Naruya Saitou and Masatoshi Nei in 1987. Usually based on DNA or protein sequence data, the algorithm requires knowledge of the distance between each pair of taxa to create the phylogenetic tree.
UPGMA is a simple agglomerative (bottom-up) hierarchical clustering method. It also has a weighted variant, WPGMA, and they are generally attributed to Sokal and Michener.
In data mining and statistics, hierarchical clustering is a method of cluster analysis that seeks to build a hierarchy of clusters. Strategies for hierarchical clustering generally fall into two categories:
Cluster analysis or clustering is the task of grouping a set of objects in such a way that objects in the same group are more similar to each other than to those in other groups (clusters). It is a main task of exploratory data analysis, and a common technique for statistical data analysis, used in many fields, including pattern recognition, image analysis, information retrieval, bioinformatics, data compression, computer graphics and machine learning.
In applied statistics, total least squares is a type of errors-in-variables regression, a least squares data modeling technique in which observational errors on both dependent and independent variables are taken into account. It is a generalization of Deming regression and also of orthogonal regression, and can be applied to both linear and non-linear models.
In mathematics, differential algebra is, broadly speaking, the area of mathematics consisting in the study of differential equations and differential operators as algebraic objects in view of deriving properties of differential equations and operators without computing the solutions, similarly as polynomial algebras are used for the study of algebraic varieties, which are solution sets of systems of polynomial equations. Weyl algebras and Lie algebras may be considered as belonging to differential algebra.
The Hungarian method is a combinatorial optimization algorithm that solves the assignment problem in polynomial time and which anticipated later primal–dual methods. It was developed and published in 1955 by Harold Kuhn, who gave it the name "Hungarian method" because the algorithm was largely based on the earlier works of two Hungarian mathematicians, Dénes Kőnig and Jenő Egerváry. However, in 2006 it was discovered that Carl Gustav Jacobi had solved the assignment problem in the 19th century, and the solution had been published posthumously in 1890 in Latin.
In solid-state physics, the tight-binding model is an approach to the calculation of electronic band structure using an approximate set of wave functions based upon superposition of wave functions for isolated atoms located at each atomic site. The method is closely related to the LCAO method used in chemistry. Tight-binding models are applied to a wide variety of solids. The model gives good qualitative results in many cases and can be combined with other models that give better results where the tight-binding model fails. Though the tight-binding model is a one-electron model, the model also provides a basis for more advanced calculations like the calculation of surface states and application to various kinds of many-body problem and quasiparticle calculations.
In statistics, single-linkage clustering is one of several methods of hierarchical clustering. It is based on grouping clusters in bottom-up fashion, at each step combining two clusters that contain the closest pair of elements not yet belonging to the same cluster as each other.
In numerical linear algebra, the alternating-direction implicit (ADI) method is an iterative method used to solve Sylvester matrix equations. It is a popular method for solving the large matrix equations that arise in systems theory and control, and can be formulated to construct solutions in a memory-efficient, factored form. It is also used to numerically solve parabolic and elliptic partial differential equations, and is a classic method used for modeling heat conduction and solving the diffusion equation in two or more dimensions. It is an example of an operator splitting method.
In mathematics, the Johnson–Lindenstrauss lemma is a result named after William B. Johnson and Joram Lindenstrauss concerning low-distortion embeddings of points from high-dimensional into low-dimensional Euclidean space. The lemma states that a set of points in a high-dimensional space can be embedded into a space of much lower dimension in such a way that distances between the points are nearly preserved. In the classical proof of the lemma, the embedding is a random orthogonal projection.
Non-linear least squares is the form of least squares analysis used to fit a set of m observations with a model that is non-linear in n unknown parameters (m ≥ n). It is used in some forms of nonlinear regression. The basis of the method is to approximate the model by a linear one and to refine the parameters by successive iterations. There are many similarities to linear least squares, but also some significant differences. In economic theory, the non-linear least squares method is applied in (i) the probit regression, (ii) threshold regression, (iii) smooth regression, (iv) logistic link regression, (v) Box–Cox transformed regressors ().
In mathematics, near sets are either spatially close or descriptively close. Spatially close sets have nonempty intersection. In other words, spatially close sets are not disjoint sets, since they always have at least one element in common. Descriptively close sets contain elements that have matching descriptions. Such sets can be either disjoint or non-disjoint sets. Spatially near sets are also descriptively near sets.
The Dunn index (DI) (introduced by J. C. Dunn in 1974) is a metric for evaluating clustering algorithms. This is part of a group of validity indices including the Davies–Bouldin index or Silhouette index, in that it is an internal evaluation scheme, where the result is based on the clustered data itself. As do all other such indices, the aim is to identify sets of clusters that are compact, with a small variance between members of the cluster, and well separated, where the means of different clusters are sufficiently far apart, as compared to the within cluster variance. For a given assignment of clusters, a higher Dunn index indicates better clustering. One of the drawbacks of using this is the computational cost as the number of clusters and dimensionality of the data increase.
Matrix completion is the task of filling in the missing entries of a partially observed matrix, which is equivalent to performing data imputation in statistics. A wide range of datasets are naturally organized in matrix form. One example is the movie-ratings matrix, as appears in the Netflix problem: Given a ratings matrix in which each entry represents the rating of movie by customer , if customer has watched movie and is otherwise missing, we would like to predict the remaining entries in order to make good recommendations to customers on what to watch next. Another example is the document-term matrix: The frequencies of words used in a collection of documents can be represented as a matrix, where each entry corresponds to the number of times the associated term appears in the indicated document.
The distributional learning theory or learning of probability distribution is a framework in computational learning theory. It has been proposed from Michael Kearns, Yishay Mansour, Dana Ron, Ronitt Rubinfeld, Robert Schapire and Linda Sellie in 1994 and it was inspired from the PAC-framework introduced by Leslie Valiant.
WPGMA is a simple agglomerative (bottom-up) hierarchical clustering method, generally attributed to Sokal and Michener.
A central problem in algorithmic graph theory is the shortest path problem. One of the generalizations of the shortest path problem is known as the single-source-shortest-paths (SSSP) problem, which consists of finding the shortest paths from a source vertex to all other vertices in the graph. There are classical sequential algorithms which solve this problem, such as Dijkstra's algorithm. In this article, however, we present two parallel algorithms solving this problem.
References
↑ Sorensen T (1948). "A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analyses of the vegetation on Danish commons". Biologiske Skrifter. 5: 1–34.
↑ Defays D (1977). "An efficient algorithm for a complete link method". The Computer Journal. 20 (4). British Computer Society: 364–366. doi:10.1093/comjnl/20.4.364.
Späth H (1980). Cluster Analysis Algorithms. Chichester: Ellis Horwood.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.