
Multiple myeloma (MM), also known as plasma cell myeloma and simply myeloma, is a cancer of plasma cells, a type of white blood cell that normally produces antibodies. Often, no symptoms are noticed initially. As it progresses, bone pain, anemia, kidney dysfunction, and infections may occur. Complications may include hypercalcemia and amyloidosis.
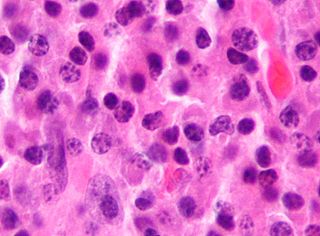
Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making aplasia, myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.

POEMS syndrome is a rare paraneoplastic syndrome caused by a clone of aberrant plasma cells. The name POEMS is an acronym for some of the disease's major signs and symptoms, as is PEP.

Cryoglobulinemia is a medical condition in which the blood contains large amounts of pathological cold sensitive antibodies called cryoglobulins – proteins that become insoluble at reduced temperatures. This should be contrasted with cold agglutinins, which cause agglutination of red blood cells.

Langerhans cell histiocytosis (LCH) is an abnormal clonal proliferation of Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes.

Monoclonal gammopathy of undetermined significance (MGUS) is a plasma cell dyscrasia in which plasma cells or other types of antibody-producing cells secrete a myeloma protein, i.e. an abnormal antibody, into the blood; this abnormal protein is usually found during standard laboratory blood or urine tests. MGUS resembles multiple myeloma and similar diseases, but the levels of antibodies are lower, the number of plasma cells in the bone marrow is lower, and it rarely has symptoms or major problems. However, since MGUS can lead to multiple myeloma, which develops at the rate of about 1.5% a year, or other symptomatic conditions, yearly monitoring is recommended.
Plasmacytoma is a plasma cell dyscrasia in which a plasma cell tumour grows within soft tissue or within the axial skeleton.

Letterer–Siwe disease, (LSD) or Abt-Letterer-Siwe disease, is one of the four recognized clinical syndromes of Langerhans cell histiocytosis (LCH) and is the most severe form, involving multiple organ systems such as the skin, bone marrow, spleen, liver, and lung. Oral cavity and gastrointestinal involvement may also be seen. LCH and all its subtypes are characterized by monoclonal migration and proliferation of specific dendritic cells.

A histiocytoma in the dog is a benign tumor. It is an abnormal growth in the skin of histiocytes (histiocytosis), a cell that is part of the immune system. A similar disease in humans, Hashimoto-Pritzker disease, is also a Langerhans cell histiocytosis. Dog breeds that may be more at risk for this tumor include Bulldogs, American Pit Bull Terriers, American Staffordshire Terriers, Scottish Terriers, Greyhounds, Boxers, and Boston Terriers. They also rarely occur in goats and cattle.

Cutis laxa or pachydermatocele is a group of rare connective tissue disorders in which the skin becomes inelastic and hangs loosely in folds.

Erdheim–Chester disease (ECD) is an extremely rare disease characterized by the abnormal multiplication of a specific type of white blood cells called histiocytes, or tissue macrophages. It was declared a histiocytic neoplasm by the World Health Organization in 2016. Onset typically is in middle age, although younger patients have been documented. The disease involves an infiltration of lipid-laden macrophages, multinucleated giant cells, an inflammatory infiltrate of lymphocytes and histiocytes in the bone marrow, and a generalized sclerosis of the long bones.
In medicine, histiocytosis is an excessive number of histiocytes, and the term is also often used to refer to a group of rare diseases which share this sign as a characteristic. Occasionally and confusingly, the term histiocytosis is sometimes used to refer to individual diseases.
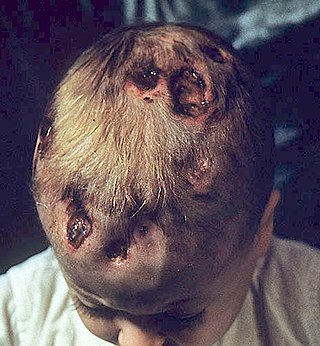
Chronic multifocal Langerhans cell histiocytosis, previously known as Hand–Schüller–Christian disease, is a type of Langerhans cell histiocytosis (LCH), which can affect multiple organs. The condition is traditionally associated with a combination of three features; bulging eyes, breakdown of bone, and diabetes insipidus, although around 75% of cases do not have all three features. Other features may include a fever and weight loss, and depending on the organs involved there may be rashes, asymmetry of the face, ear infections, signs in the mouth and the appearance of advanced gum disease. Features relating to lung and liver disease may occur.

Rosai–Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy or sometimes as Destombes–Rosai–Dorfman disease, is a rare disorder of unknown cause that is characterized by abundant histiocytes in the lymph nodes or other locations throughout the body.
In hematology, plasma cell dyscrasias are a spectrum of progressively more severe monoclonal gammopathies in which a clone or multiple clones of pre-malignant or malignant plasma cells over-produce and secrete into the blood stream a myeloma protein, i.e. an abnormal monoclonal antibody or portion thereof. The exception to this rule is the disorder termed non-secretory multiple myeloma; this disorder is a form of plasma cell dyscrasia in which no myeloma protein is detected in serum or urine of individuals who have clear evidence of an increase in clonal bone marrow plasma cells and/or evidence of clonal plasma cell-mediated tissue injury. Here, a clone of plasma cells refers to group of plasma cells that are abnormal in that they have an identical genetic identity and therefore are descendants of a single genetically distinct ancestor cell.
Free light chains (FLCs) are immunoglobulin light chains that are found in the serum (blood) in an unbound (free) state. In recent decades, measuring the amount of free light chains (FLCs) in the blood has become a practical clinical test. FLC tests can be used to diagnose and monitor diseases like multiple myeloma and amyloidosis.

Light chain deposition disease (LCDD) is a rare blood cell disease which is characterized by deposition of fragments of infection-fighting immunoglobulins, called light chains (LCs), in the body. LCs are normally cleared by the kidneys, but in LCDD, these light chain deposits damage organs and cause disease. The kidneys are almost always affected and this often leads to kidney failure. About half of people with light chain deposition disease also have a plasma cell dyscrasia, a spectrum of diseases that includes multiple myeloma, Waldenström's macroglobulinemia, and the monoclonal gammopathy of undetermined significance premalignant stages of these two diseases. Unlike in AL amyloidosis, in which light chains are laid down in characteristic amyloid deposits, in LCDD, light chains are deposited in non-amyloid granules.
The xanthogranulomatous process (XP), is a form of acute and chronic inflammation characterized by an exuberant clustering of foamy macrophages among other inflammatory cells. Localization in the kidney and renal pelvis has been the most frequent and better known occurrence followed by that in the gallbladder but many others have been subsequently recorded. The pathological findings of the process and etiopathogenetic and clinical observations have been reviewed by Cozzutto and Carbone.
Monoclonal immunoglobulin deposition disease, or MIDD, is a disease characterised by the deposition of monoclonal immunoglobulins on the basement membrane of the kidney. Monoclonal immunoglobulins are produced by monoclonal plasma cells, which are found in a variety of plasma cell dyscrasias. The deposition of monoclonal immunoglobulins on the basement membrane of the kidney causes renal impairment. As well as the kidney, MIDD may also affect the liver, heart, peripheral nerves, lung and skin.
Monoclonal gammopathy of renal significance (MGRS) are a group of kidney disorders that present with kidney damage due to nephrotoxic monoclonal immunoglobulins secreted by clonal plasma cells or B cells. By definition, people with MGRS do not meet criteria for multiple myeloma or other hematologic malignancies. The term MGRS was introduced in 2012 by the International Kidney and Monoclonal Gammopathy Research Group (IKMG). MGRS is associated with monoclonal gammopathy of undetermined significance (MGUS). People with MGUS have a monoclonal gammopathy but does not meet the criteria for the clonal burden nor the presence of end organ damage seen in hematologic malignancies. In a population based study based on the NHANES III health survey; 6% of patients with MGUS were subsequently classified as having MGRS. The prevalence and incidence of MGRS in the general population or in specific populations is not known but it is more prevalent in those over the age of 50 as there is a monoclonal protein (M-protein) present in 3% of those 50 and years older and 5% of those 70 years and older, placing those 50 and older at increased risk of MGRS.

