Related Research Articles

Dermatofibrosarcoma protuberans (DFSP) is a rare locally aggressive malignant cutaneous soft-tissue sarcoma. DFSP develops in the connective tissue cells in the middle layer of the skin (dermis). Estimates of the overall occurrence of DFSP in the United States are 0.8 to 4.5 cases per million persons per year. In the United States, DFSP accounts for between 1 and 6 percent of all soft-tissue sarcomas and 18 percent of all cutaneous soft-tissue sarcomas. In the Surveillance, Epidemiology and End Results (SEER) tumor registry from 1992 through 2004, DFSP was second only to Kaposi sarcoma.

Liposarcomas are the most common subtype of soft tissue sarcomas, accounting for at least 20% of all sarcomas in adults. Soft tissue sarcomas are rare neoplasms with over 150 different histological subtypes or forms. Liposarcomas arise from the precursor lipoblasts of the adipocytes in adipose tissues. Adipose tissues are distributed throughout the body, including such sites as the deep and more superficial layers of subcutaneous tissues as well as in less surgically accessible sites like the retroperitoneum and visceral fat inside the abdominal cavity.

Fibrosarcoma is a malignant mesenchymal tumour derived from fibrous connective tissue and characterized by the presence of immature proliferating fibroblasts or undifferentiated anaplastic spindle cells in a storiform pattern. Fibrosarcomas mainly arise in people between the ages of 25 and 79. It originates in fibrous tissues of the bone and invades long or flat bones such as the femur, tibia, and mandible. It also involves the periosteum and overlying muscle.

Nodular fasciitis (NF) is a benign, soft tissue tumor composed of myofibroblasts that typically occurs in subcutaneous tissue, fascia, and/or muscles. The literature sometimes titles rare NF variants according to their tissue locations. The most frequently used and important of these are cranial fasciitis and intravascular fasciitis. In 2020, the World Health Organization classified nodular fasciitis as in the category of benign fibroblastic/myofibroblastic tumors. NF is the most common of the benign fibroblastic proliferative tumors of soft tissue.
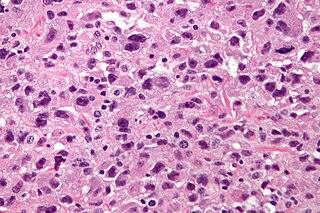
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

ETV6 protein is a transcription factor that in humans is encoded by the ETV6 gene. The ETV6 protein regulates the development and growth of diverse cell types, particularly those of hematological tissues. However, its gene, ETV6 frequently suffers various mutations that lead to an array of potentially lethal cancers, i.e., ETV6 is a clinically significant proto-oncogene in that it can fuse with other genes to drive the development and/or progression of certain cancers. However, ETV6 is also an anti-oncogene or tumor suppressor gene in that mutations in it that encode for a truncated and therefore inactive protein are also associated with certain types of cancers.

Congenital mesoblastic nephroma, while rare, is the most common kidney neoplasm diagnosed in the first three months of life and accounts for 3-5% of all childhood renal neoplasms. This neoplasm is generally non-aggressive and amenable to surgical removal. However, a readily identifiable subset of these kidney tumors has a more malignant potential and is capable of causing life-threatening metastases. Congenital mesoblastic nephroma was first named as such in 1967 but was recognized decades before this as fetal renal hamartoma or leiomyomatous renal hamartoma.
Giant cell fibroblastoma (GCF) is a rare type of soft-tissue tumor marked by painless nodules in the dermis and subcutaneous tissue. These tumors may come back after surgery, but they do not spread to other parts of the body. They occur mostly in boys. GCF tumor tissues consist of bland spindle-shaped or stellate-shaped cells interspersed among multinucleated giant cells.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first definitively characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a cluster of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Less commonly, cases are reported in the pelvis, vulva, penis, and spine.
Porocarcinoma (PCA) is a rare form of skin cancer that develops in eccrine sweat glands, i.e. the body's widely distributed major type of sweat glands, as opposed to the apocrine sweat glands which are located primarily in the armpits and perineal area. This cancer typically develops in individuals as a single cutaneous tumor in the intraepidermal spiral part of these sweat glands' ducts at or near to where they open on the skin's surface. PCA tumors are classified as one form of the cutaneous adnexal tumors; in a study of 2,205 cases, PCA was the most common (11.8%) form of these tumors.

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.

Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of the mesodermal cells that form the connective tissues which support virtually all of the organs and tissues of the body. IMT was formerly termed inflammatory pseudotumor. Currently, however, inflammatory pseudotumor designates a large and heterogeneous group of soft tissue tumors that includes inflammatory myofibroblastic tumor, plasma cell granuloma, xanthomatous pseudotumor, solitary mast cell granuloma, inflammatory fibrosarcoma, pseudosarcomatous myofibroblastic proliferation, myofibroblastoma, inflammatory myofibrohistiocytic proliferation, and other tumors that develop from connective tissue cells. Inflammatory pseudotumour is a generic term applied to various neoplastic and non-neoplastic tissue lesions which share a common microscopic appearance consisting of spindle cells and a prominent presence of the white blood cells that populate chronic or, less commonly, acute inflamed tissues.
Mammary analogue secretory carcinoma (MASC), also termed MASCSG, is a salivary gland neoplasm. It is a secretory carcinoma which shares the microscopic pathologic features with other types of secretory carcinomas including mammary secretory carcinoma, secretory carcinoma of the skin, and salivary gland–type carcinoma of the thyroid. MASCSG was first described by Skálová et al. in 2010. The authors of this report found a chromosome translocation in certain salivary gland tumors, i.e. a (12;15)(p13;q25) fusion gene mutation. The other secretory carcinoma types carry this fusion gene.
Acral myxoinflammatory fibroblastic sarcoma (AMSF), also termed myxoinflammatory fibroblastic sarcoma (MSF), is a rare, low-grade, soft tissue tumor that the World Health Organization (2020) classified as in the category of rarely metastasizing fibroblastic and myofibroblastic tumors. It is a locally aggressive neoplasm that often recurs at the site of its surgical removal. However, it usually grows slowly and in only 1–2% of cases spreads to distant tissues.

Proliferative fasciitis and proliferative myositis (PF/PM) are rare benign soft tissue lesions that increase in size over several weeks and often regress over the ensuing 1–3 months. The lesions in PF/PM are typically obvious tumors or swellings. Historically, many studies had grouped the two descriptive forms of PF/PM as similar disorders with the exception that proliferative fasciitis occurs in subcutaneous tissues while proliferative myositis occurs in muscle tissues. In 2020, the World Health Organization agreed with this view and defined these lesions as virtually identical disorders termed proliferative fasciitis/proliferative myositis or proliferative fasciitis and proliferative myositis. The Organization also classified them as one of the various forms of the fibroblastic and myofibroblastic tumors.
Lipofibromatosis (LPF) is an extremely rare soft tissue tumor which was first clearly described in 2000 by Fetsch et al as a strictly pediatric, locally invasive, and often recurrent tumor. It is nonetheless a non-metastasizing, i.e. benign, tumor. While even the more recent literature has sometimes regarded LPF as a strictly childhood disorder, rare cases of LPF has been diagnosed in adults. The diagnosis of lipofibromatosis should not be automatically discarded because of an individual's age.
Fibroblastic and myofibroblastic tumors (FMTs) develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
Lipofibromatosis-like neural tumor (LPF-NT) is an extremely rare soft tissue tumor first described by Agaram et al in 2016. As of mid-2021, at least 39 cases of LPF-NT have been reported in the literature. LPF-NT tumors have several features that resemble lipofibromatosis (LPF) tumors, malignant peripheral nerve sheath tumors, spindle cell sarcomas, low-grade neural tumors, peripheral nerve sheath tumors, and other less clearly defined tumors; Prior to the Agaram at al report, LPF-NTs were likely diagnosed as variants or atypical forms of these tumors. The analyses of Agaram at al and subsequent studies uncovered critical differences between LPF-NT and the other tumor forms which suggest that it is a distinct tumor entity differing not only from lipofibromatosis but also the other tumor forms.
Sclerosing epithelioid fibrosarcoma (SEF) is a very rare malignant tumor of soft tissues that on microscopic examination consists of small round or ovoid neoplastic epithelioid fibroblast-like cells, i.e. cells that have features resembling both epithelioid cells and fibroblasts. In 2020, the World Health Organization classified SEF as a distinct tumor type in the category of malignant fibroblastic and myofibroblastic tumors. However, current studies have reported that low-grade fibromyxoid sarcoma (LGFMS) has many clinically and pathologically important features characteristic of SEF; these studies suggest that LGSFMS may be an early form of, and over time progress to become, a SEF. Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF, SEF and LGFMS are here regarded as different tumor forms.
Angiofibroma of soft tissue (AFST), also termed angiofibroma, not otherwise specified, is a recently recognized and rare disorder that was classified in the category of benign fibroblastic and myofibroblastic tumors by the World Health Organization in 2020. An AFST tumor is a neoplasm that was first described by A. Mariño-Enríquez and C.D. Fletcher in 2012.
References
- 1 2 3 4 5 6 Baranov E, Hornick JL (March 2020). "Soft Tissue Special Issue: Fibroblastic and Myofibroblastic Neoplasms of the Head and Neck". Head and Neck Pathology. 14 (1): 43–58. doi:10.1007/s12105-019-01104-3. PMC 7021862 . PMID 31950474.
- 1 2 3 4 5 Verma H, Sehgal K, Panchal KB, Chakraborty S, Biswas B, Mukherjee G, Midha D, Biswas G (March 2020). "Presentation and Management of Dermatofibrosarcoma Protuberans: a Single Center Protocol". Indian Journal of Surgical Oncology. 11 (1): 35–40. doi:10.1007/s13193-019-01007-3. PMC 7064730 . PMID 32205967.
- 1 2 3 4 Liang CA, Jambusaria-Pahlajani A, Karia PS, Elenitsas R, Zhang PD, Schmults CD (October 2014). "A systematic review of outcome data for dermatofibrosarcoma protuberans with and without fibrosarcomatous change". Journal of the American Academy of Dermatology. 71 (4): 781–6. doi:10.1016/j.jaad.2014.03.018. PMID 24755121.
- ↑ Braswell DS, Ayoubi N, Motaparthi K, Walker A (April 2020). "Dermatofibrosarcoma protuberans with features of giant cell fibroblastoma in an adult". Journal of Cutaneous Pathology. 47 (4): 317–320. doi:10.1111/cup.13601. PMID 32163628. S2CID 212691248.
- ↑ Jha P, Moosavi C, Fanburg-Smith JC (April 2007). "Giant cell fibroblastoma: an update and addition of 86 new cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz M. Enzinger". Annals of Diagnostic Pathology. 11 (2): 81–8. doi:10.1016/j.anndiagpath.2006.12.010. PMID 17349565.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394 . PMID 33179614.
- 1 2 3 4 5 Mentzel T, Beham A, Katenkamp D, Dei Tos AP, Fletcher CD (May 1998). "Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance". The American Journal of Surgical Pathology. 22 (5): 576–87. doi:10.1097/00000478-199805000-00009. PMID 9591728.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Hao X, Billings SD, Wu F, Stultz TW, Procop GW, Mirkin G, Vidimos AT (June 2020). "Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment". Journal of Clinical Medicine. 9 (6): 1752. doi: 10.3390/jcm9061752 . PMC 7355835 . PMID 32516921.
- 1 2 3 Li Y, Liang J, Xu X, Jiang X, Wang C, Chen S, Xiang B, Ji Y (January 2021). "Clinicopathological features of fibrosarcomatous dermatofibrosarcoma protuberans and the construction of a back-propagation neural network recognition model". Orphanet Journal of Rare Diseases. 16 (1): 48. doi: 10.1186/s13023-021-01698-4 . PMC 7836157 . PMID 33499900.
- ↑ Qu Q, Xuan W, Fan GH (January 2015). "Roles of resolvins in the resolution of acute inflammation". Cell Biology International. 39 (1): 3–22. doi:10.1002/cbin.10345. PMID 25052386. S2CID 10160642.
- 1 2 Chen Y, Shi YZ, Feng XH, Wang XT, He XL, Zhao M (July 2021). "Novel TNC-PDGFD fusion in fibrosarcomatous dermatofibrosarcoma protuberans: a case report". Diagnostic Pathology. 16 (1): 63. doi: 10.1186/s13000-021-01123-1 . PMC 8276425 . PMID 34256767.
- 1 2 Abbott JJ, Erickson-Johnson M, Wang X, Nascimento AG, Oliveira AM (November 2006). "Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Modern Pathology. 19 (11): 1512–8. doi:10.1038/modpathol.3800695. PMID 16980946. S2CID 276608.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Allen A, Ahn C, Sangüeza OP (October 2019). "Dermatofibrosarcoma Protuberans". Dermatologic Clinics. 37 (4): 483–488. doi:10.1016/j.det.2019.05.006. PMID 31466588. S2CID 201672710.
- ↑ Terrier-Lacombe MJ, Guillou L, Maire G, Terrier P, Vince DR, de Saint Aubain Somerhausen N, Collin F, Pedeutour F, Coindre JM (January 2003). "Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data--a study from the French Federation of Cancer Centers Sarcoma Group". The American Journal of Surgical Pathology. 27 (1): 27–39. doi:10.1097/00000478-200301000-00004. PMID 12502925. S2CID 34359313.
- ↑ Chicaud M, Frassati-Biaggi A, Kaltenbach S, Karanian M, Orbach D, Fraitag S (January 2021). "Dermatofibrosarcoma protuberans, fibrosarcomatous variant: A rare tumor in children". Pediatric Dermatology. 38 (1): 217–222. doi:10.1111/pde.14393. PMID 33010051. S2CID 222159613.
- 1 2 3 4 5 Erdem O, Wyatt AJ, Lin E, Wang X, Prieto VG (February 2012). "Dermatofibrosarcoma protuberans treated with wide local excision and followed at a cancer hospital: prognostic significance of clinicopathologic variables". The American Journal of Dermatopathology. 34 (1): 24–34. doi:10.1097/DAD.0b013e3182120671. PMID 21785324. S2CID 1402786.
- 1 2 3 4 5 Kim J, Yasuda M, Suto M, Kishi C, Motegi SI, Okamoto M, Ishikawa O (December 2019). "Unresectable local recurrence of dermatofibrosarcoma protuberans with fibrosarcomatous change treated with carbon-ion radiotherapy after neoadjuvant chemotherapy". The Journal of Dermatology. 46 (12): e457–e458. doi:10.1111/1346-8138.15056. PMID 31435947. S2CID 201274977.
- 1 2 3 4 5 Miyagawa T, Kadono T, Kimura T, Saigusa R, Yoshizaki A, Miyagaki T, Yamada D, Masui Y, Fujita H, Sato S (March 2017). "Pazopanib induced a partial response in a patient with metastatic fibrosarcomatous dermatofibrosarcoma protuberans without genetic translocations resistant to mesna, doxorubicin, ifosfamide and dacarbazine chemotherapy and gemcitabine-docetaxel chemotherapy". The Journal of Dermatology. 44 (3): e21–e22. doi:10.1111/1346-8138.13717. PMID 27988943. S2CID 7604809.
- 1 2 Tsuchihashi K, Kusaba H, Yamada Y, Okumura Y, Shimokawa H, Komoda M, Uchino K, Yoshihiro T, Tsuruta N, Hanamura F, Inadomi K, Ito M, Sagara K, Nakano M, Nio K, Arita S, Ariyama H, Kohashi K, Tominaga R, Oda Y, Akashi K, Baba E (May 2017). "Programmed death-ligand 1 expression is associated with fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Molecular and Clinical Oncology. 6 (5): 665–668. doi:10.3892/mco.2017.1197. PMC 5431145 . PMID 28515919.
- 1 2 3 4 5 Saiag P, Grob JJ, Lebbe C, Malvehy J, del Marmol V, Pehamberger H, Peris K, Stratigos A, Middelton M, Basholt L, Testori A, Garbe C (November 2015). "Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline". European Journal of Cancer. 51 (17): 2604–8. doi:10.1016/j.ejca.2015.06.108. PMID 26189684.
- ↑ "COL1A1 collagen type I alpha 1 chain [Homo sapiens (Human)] - Gene - NCBI".
- ↑ Hiraki-Hotokebuchi Y, Yamada Y, Kohashi K, Yamamoto H, Endo M, Setsu N, Yuki K, Ito T, Iwamoto Y, Furue M, Oda Y (September 2017). "Alteration of PDGFRβ-Akt-mTOR pathway signaling in fibrosarcomatous transformation of dermatofibrosarcoma protuberans". Human Pathology. 67: 60–68. doi:10.1016/j.humpath.2017.07.001. hdl: 2324/2348723 . PMID 28711648.
- ↑ "EMILIN2 elastin microfibril interfacer 2 [Homo sapiens (Human)] - Gene - NCBI".
- ↑ Nakamura I, Kariya Y, Okada E, Yasuda M, Matori S, Ishikawa O, Uezato H, Takahashi K (December 2015). "A Novel Chromosomal Translocation Associated With COL1A2-PDGFB Gene Fusion in Dermatofibrosarcoma Protuberans: PDGF Expression as a New Diagnostic Tool". JAMA Dermatology. 151 (12): 1330–1337. doi: 10.1001/jamadermatol.2015.2389 . PMID 26332510.
- ↑ Ratan R, Patel SR (October 2016). "Chemotherapy for soft tissue sarcoma". Cancer. 122 (19): 2952–60. doi: 10.1002/cncr.30191 . PMID 27434055. S2CID 3609199.
- ↑ Lyu A, Wang Q (August 2018). "Dermatofibrosarcoma protuberans: A clinical analysis". Oncology Letters. 16 (2): 1855–1862. doi:10.3892/ol.2018.8802. PMC 6036409 . PMID 30008876.