In a chemical reaction, chemical equilibrium is the state in which both the reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. This state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but they are equal. Thus, there are no net changes in the concentrations of the reactants and products. Such a state is known as dynamic equilibrium.
Raoult's law ( law) is a relation of physical chemistry, with implications in thermodynamics. Proposed by French chemist François-Marie Raoult in 1887, it states that the partial pressure of each component of an ideal mixture of liquids is equal to the vapor pressure of the pure component multiplied by its mole fraction in the mixture. In consequence, the relative lowering of vapor pressure of a dilute solution of nonvolatile solute is equal to the mole fraction of solute in the solution.
In electrochemistry, the Nernst equation is a chemical thermodynamical relationship that permits the calculation of the reduction potential of a reaction from the standard electrode potential, absolute temperature, the number of electrons involved in the redox reaction, and activities of the chemical species undergoing reduction and oxidation respectively. It was named after Walther Nernst, a German physical chemist who formulated the equation.

A eutectic system or eutectic mixture is a homogeneous mixture that has a melting point lower than those of the constituents. The lowest possible melting point over all of the mixing ratios of the constituents is called the eutectic temperature. On a phase diagram, the eutectic temperature is seen as the eutectic point.
In chemical thermodynamics, activity is a measure of the "effective concentration" of a species in a mixture, in the sense that the species' chemical potential depends on the activity of a real solution in the same way that it would depend on concentration for an ideal solution. The term "activity" in this sense was coined by the American chemist Gilbert N. Lewis in 1907.
In thermodynamics, the chemical potential of a species is the energy that can be absorbed or released due to a change of the particle number of the given species, e.g. in a chemical reaction or phase transition. The chemical potential of a species in a mixture is defined as the rate of change of free energy of a thermodynamic system with respect to the change in the number of atoms or molecules of the species that are added to the system. Thus, it is the partial derivative of the free energy with respect to the amount of the species, all other species' concentrations in the mixture remaining constant. When both temperature and pressure are held constant, and the number of particles is expressed in moles, the chemical potential is the partial molar Gibbs free energy. At chemical equilibrium or in phase equilibrium, the total sum of the product of chemical potentials and stoichiometric coefficients is zero, as the free energy is at a minimum. In a system in diffusion equilibrium, the chemical potential of any chemical species is uniformly the same everywhere throughout the system.
In physical chemistry, Henry's law is a gas law that states that the amount of dissolved gas in a liquid is directly proportional to its partial pressure above the liquid. The proportionality factor is called Henry's law constant. It was formulated by the English chemist William Henry, who studied the topic in the early 19th century.
In thermochemistry, the enthalpy of solution is the enthalpy change associated with the dissolution of a substance in a solvent at constant pressure resulting in infinite dilution.
In chemistry, an ideal solution or ideal mixture is a solution that exhibits thermodynamic properties analogous to those of a mixture of ideal gases. The enthalpy of mixing is zero as is the volume change on mixing by definition; the closer to zero the enthalpy of mixing is, the more "ideal" the behavior of the solution becomes. The vapor pressures of the solvent and solute obey Raoult's law and Henry's law, respectively, and the activity coefficient is equal to one for each component.
The equilibrium constant of a chemical reaction is the value of its reaction quotient at chemical equilibrium, a state approached by a dynamic chemical system after sufficient time has elapsed at which its composition has no measurable tendency towards further change. For a given set of reaction conditions, the equilibrium constant is independent of the initial analytical concentrations of the reactant and product species in the mixture. Thus, given the initial composition of a system, known equilibrium constant values can be used to determine the composition of the system at equilibrium. However, reaction parameters like temperature, solvent, and ionic strength may all influence the value of the equilibrium constant.
In chemical thermodynamics, the fugacity of a real gas is an effective partial pressure which replaces the mechanical partial pressure in an accurate computation of chemical equilibrium. It is equal to the pressure of an ideal gas which has the same temperature and molar Gibbs free energy as the real gas.
In thermodynamics, an activity coefficient is a factor used to account for deviation of a mixture of chemical substances from ideal behaviour. In an ideal mixture, the microscopic interactions between each pair of chemical species are the same and, as a result, properties of the mixtures can be expressed directly in terms of simple concentrations or partial pressures of the substances present e.g. Raoult's law. Deviations from ideality are accommodated by modifying the concentration by an activity coefficient. Analogously, expressions involving gases can be adjusted for non-ideality by scaling partial pressures by a fugacity coefficient.
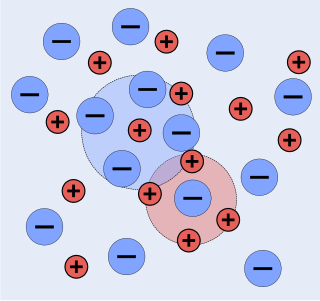
The chemists Peter Debye and Erich Hückel noticed that solutions that contain ionic solutes do not behave ideally even at very low concentrations. So, while the concentration of the solutes is fundamental to the calculation of the dynamics of a solution, they theorized that an extra factor that they termed gamma is necessary to the calculation of the activities of the solution. Hence they developed the Debye–Hückel equation and Debye–Hückel limiting law. The activity is only proportional to the concentration and is altered by a factor known as the activity coefficient . This factor takes into account the interaction energy of ions in solution.

The non-random two-liquid model is an activity coefficient model that correlates the activity coefficients of a compound with its mole fractions in the liquid phase concerned. It is frequently applied in the field of chemical engineering to calculate phase equilibria. The concept of NRTL is based on the hypothesis of Wilson that the local concentration around a molecule is different from the bulk concentration. This difference is due to a difference between the interaction energy of the central molecule with the molecules of its own kind and that with the molecules of the other kind . The energy difference also introduces a non-randomness at the local molecular level. The NRTL model belongs to the so-called local-composition models. Other models of this type are the Wilson model, the UNIQUAC model, and the group contribution model UNIFAC. These local-composition models are not thermodynamically consistent for a one-fluid model for a real mixture due to the assumption that the local composition around molecule i is independent of the local composition around molecule j. This assumption is not true, as was shown by Flemr in 1976. However, they are consistent if a hypothetical two-liquid model is used.
In thermodynamics, the enthalpy of mixing is the enthalpy liberated or absorbed from a substance upon mixing. When a substance or compound is combined with any other substance or compound, the enthalpy of mixing is the consequence of the new interactions between the two substances or compounds. This enthalpy, if released exothermically, can in an extreme case cause an explosion.

In statistical thermodynamics, UNIQUAC is an activity coefficient model used in description of phase equilibria. The model is a so-called lattice model and has been derived from a first order approximation of interacting molecule surfaces. The model is, however, not fully thermodynamically consistent due to its two-liquid mixture approach. In this approach the local concentration around one central molecule is assumed to be independent from the local composition around another type of molecule.
In thermodynamics, a partial molar property is a quantity which describes the variation of an extensive property of a solution or mixture with changes in the molar composition of the mixture at constant temperature and pressure. It is the partial derivative of the extensive property with respect to the amount of the component of interest. Every extensive property of a mixture has a corresponding partial molar property.
An osmotic coefficient is a quantity which characterises the deviation of a solvent from ideal behaviour, referenced to Raoult's law. It can be also applied to solutes. Its definition depends on the ways of expressing chemical composition of mixtures.
In thermodynamics, an apparent molar property of a solution component in a mixture or solution is a quantity defined with the purpose of isolating the contribution of each component to the non-ideality of the mixture. It shows the change in the corresponding solution property per mole of that component added, when all of that component is added to the solution. It is described as apparent because it appears to represent the molar property of that component in solution, provided that the properties of the other solution components are assumed to remain constant during the addition. However this assumption is often not justified, since the values of apparent molar properties of a component may be quite different from its molar properties in the pure state.
The shear viscosity of a fluid is a material property that describes the friction between internal neighboring fluid surfaces flowing with different fluid velocities. This friction is the effect of (linear) momentum exchange caused by molecules with sufficient energy to move between these fluid sheets due to fluctuations in their motion. The viscosity is not a material constant, but a material property that depends on temperature, pressure, fluid mixture composition, local velocity variations. This functional relationship is described by a mathematical viscosity model called a constitutive equation which is usually far more complex than the defining equation of shear viscosity. One such complicating feature is the relation between the viscosity model for a pure fluid and the model for a fluid mixture which is called mixing rules. When scientists and engineers use new arguments or theories to develop a new viscosity model, instead of improving the reigning model, it may lead to the first model in a new class of models. This article will display one or two representative models for different classes of viscosity models, and these classes are:

























