Related Research Articles

A unibrow is a single eyebrow created when the two eyebrows meet in the middle above the bridge of the nose. The hair above the bridge of the nose is of the same color and thickness as the eyebrows, such that they converge to form one uninterrupted line of hair.
Neurodevelopmental disorders are a group of conditions that begin to emerge during childhood. According to the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, (DSM-5) published in 2013, these conditions generally appear in early childhood, usually before children start school, and can persist into adulthood. The key characteristic of all these disorders is that they negatively impact a person's functioning in one or more domains of life depending on the disorder and deficits it has caused. All of these disorders and their levels of impairment exist on a spectrum, and affected individuals can experience varying degrees of symptoms and deficits, despite having the same diagnosis.
D-Bifunctional protein deficiency is an autosomal recessive peroxisomal fatty acid oxidation disorder. Peroxisomal disorders are usually caused by a combination of peroxisomal assembly defects or by deficiencies of specific peroxisomal enzymes. The peroxisome is an organelle in the cell similar to the lysosome that functions to detoxify the cell. Peroxisomes contain many different enzymes, such as catalase, and their main function is to neutralize free radicals and detoxify drugs. For this reason peroxisomes are ubiquitous in the liver and kidney. D-BP deficiency is the most severe peroxisomal disorder, often resembling Zellweger syndrome.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Kohlschütter–Tönz syndrome (KTS), also called amelo-cerebro-hypohidrotic syndrome, is a rare inherited syndrome characterized by epilepsy, psychomotor delay or regression, intellectual disability, and yellow teeth caused by amelogenesis imperfecta. It is a type A ectodermal dysplasia.
Xia-Gibbs syndrome, is genetic disorder caused by a heterozygous mutation in the AHDC1 gene on chromosome 1p36.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.
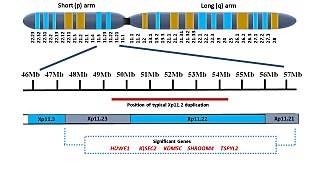
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
SYT1-associated neurodevelopmental disorder, also known as Baker-Gordon syndrome, is a rare genetic disorder caused by mutations in the synaptotagmin-1 (SYT1) gene.
ADNP syndrome, also known as Helsmoortel-Van der Aa syndrome (HVDAS), is a non-inherited neurodevelopmental disorder caused by mutations in the activity-dependent neuroprotector homeobox (ADNP) gene.
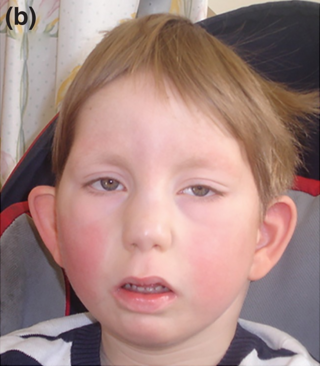
Okamoto syndrome (OS), also known as Au–Kline syndrome (AKS), is a very rare autosomal dominant genetic condition characterised by congenital hydronephrosis, low muscle tone, heart defects, intellectual disability and characteristic facial features. Those affected often have neurological and skeletal abnormalities, as well as frequent urinary tract infections. Language and walking are usually delayed. Facial features include prominent, downturned ears, an open, downturned mouth and drooping eyelids (ptosis).
Jordan's syndrome (JS) or PPP2R5D-related intellectual disability is a rare autosomal dominant neurodevelopmental disorder caused by de novo mutations in the PPP2R5D gene. It is characterized by hypotonia, intellectual disability, and macrocephaly. Children with JS may also have epilepsy or meet criteria for diagnosis with autism spectrum disorder.

PURA syndrome, also known as PURA-related neurodevelopmental disorder, is a rare novel genetic disorder which is characterized by developmental and speech delay, neo-natal hypotonia, failure to thrive, excessive sleepiness, epilepsy, and other anomalies.
CHAMP1-associated intellectual disability syndrome, also known as autosomal dominant intellectual disability type 40, is a rare genetic disorder characterized by intellectual disabilities, developmental delays, facial dysmorphisms, and other anomalies.
Autosomal dominant intellectual disability-craniofacial anomalies-cardiac defects syndrome is a rare genetic disorder which is characterized by multi-systemic symptoms primarily affecting the intellect and post-natal development.
GRIN2B-related neurodevelopmental disorder is a rare neurodevelopmental disorder which is characterized by developmental delays and intellectual disabilities of variable degrees, muscle tone anomalies, feeding difficulties, and behavioral problems.

Snijders Blok–Campeau syndrome is a genetic disorder caused by mutations in the CHD3 gene. It is characterized by impaired intellectual development, macrocephaly, dysarthria and apraxia of speech, and certain distinctive facial features.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
Salt and pepper developmental regression syndrome, also known as Amish infantile epileptic syndrome or GM3 deficiency syndrome, is a rare autosomal recessive progressive neurological disorder characterized by developmental delay, severe intellectual disability, seizures, and skin pigmentation irregularities. The clinical symptoms of this condition start manifesting soon after birth, during the newborn/neo-natal stage of life.
TRPM3-related neurodevelopmental disorder is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. The broad phenotype includes global developmental delay, intellectual disability, epilepsy, musculoskeletal anomalies, altered pain perception, ataxia, hypotonia, nystagmus, and cerebellar atrophy.
References
- ↑ "Researchers discover new neurodevelopmental disorder". ScienceDaily. Retrieved 2022-06-21.
- ↑ Yeh, Chien-Hung; Bellon, Marcia; Nicot, Christophe (2018-08-07). "FBXW7: a critical tumor suppressor of human cancers". Molecular Cancer. 17 (1): 115. doi: 10.1186/s12943-018-0857-2 . ISSN 1476-4598. PMC 6081812 . PMID 30086763.
- ↑ Stephenson, Sarah E. M.; Costain, Gregory; Blok, Laura E. R.; Silk, Michael A.; Nguyen, Thanh Binh; Dong, Xiaomin; Alhuzaimi, Dana E.; Dowling, James J.; Walker, Susan; Amburgey, Kimberly; Hayeems, Robin Z. (2022-04-07). "Germline variants in tumor suppressor FBXW7 lead to impaired ubiquitination and a neurodevelopmental syndrome". American Journal of Human Genetics. 109 (4): 601–617. doi:10.1016/j.ajhg.2022.03.002. ISSN 1537-6605. PMC 9069070 . PMID 35395208.
- ↑ "New neurodevelopmental disorder identified". www.labonline.com.au. Retrieved 2022-06-21.[ permanent dead link ]
- ↑ April 8, Karishma Abhishek on; AM, 2022 at 12:05 (8 April 2022). "Discovery of New Neurodevelopmental Syndrome Linked to Tumor-specific Genes". Medindia. Retrieved 2022-06-21.
{{cite web}}: CS1 maint: numeric names: authors list (link) - ↑ "Redirecting". linkinghub.elsevier.com. Retrieved 2022-06-21.