Spontaneous emission is the process in which a quantum mechanical system transits from an excited energy state to a lower energy state and emits a quantized amount of energy in the form of a photon. Spontaneous emission is ultimately responsible for most of the light we see all around us; it is so ubiquitous that there are many names given to what is essentially the same process. If atoms are excited by some means other than heating, the spontaneous emission is called luminescence. For example, fireflies are luminescent. And there are different forms of luminescence depending on how excited atoms are produced. If the excitation is effected by the absorption of radiation the spontaneous emission is called fluorescence. Sometimes molecules have a metastable level and continue to fluoresce long after the exciting radiation is turned off; this is called phosphorescence. Figurines that glow in the dark are phosphorescent. Lasers start via spontaneous emission, then during continuous operation work by stimulated emission.

A quantum mechanical system or particle that is bound—that is, confined spatially—can only take on certain discrete values of energy, called energy levels. This contrasts with classical particles, which can have any amount of energy. The term is commonly used for the energy levels of the electrons in atoms, ions, or molecules, which are bound by the electric field of the nucleus, but can also refer to energy levels of nuclei or vibrational or rotational energy levels in molecules. The energy spectrum of a system with such discrete energy levels is said to be quantized.
In quantum chemistry and molecular physics, the Born–Oppenheimer (BO) approximation is the best-known mathematical approximation in molecular dynamics. Specifically, it is the assumption that the wave functions of atomic nuclei and electrons in a molecule can be treated separately, based on the fact that the nuclei are much heavier than the electrons. Due to the larger relative mass of a nucleus compared to an electron, the coordinates of the nuclei in a system are approximated as fixed, while the coordinates of the electrons are dynamic. The approach is named after Max Born and his 23-year-old graduate student J. Robert Oppenheimer, the latter of whom proposed it in 1927 during a period of intense ferment in the development of quantum mechanics.
Density functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals - that is, functions that accept a function as input and output a single real number. In the case of DFT, these are functionals of the spatially dependent electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.

The Stark effect is the shifting and splitting of spectral lines of atoms and molecules due to the presence of an external electric field. It is the electric-field analogue of the Zeeman effect, where a spectral line is split into several components due to the presence of the magnetic field. Although initially coined for the static case, it is also used in the wider context to describe the effect of time-dependent electric fields. In particular, the Stark effect is responsible for the pressure broadening of spectral lines by charged particles in plasmas. For most spectral lines, the Stark effect is either linear or quadratic with a high accuracy.
In condensed matter physics, scintillation is the physical process where a material, called a scintillator, emits ultraviolet or visible light under excitation from high energy photons or energetic particles. See scintillator and scintillation counter for practical applications.
The adiabatic theorem is a concept in quantum mechanics. Its original form, due to Max Born and Vladimir Fock (1928), was stated as follows:
In quantum mechanics, a rotational transition is an abrupt change in angular momentum. Like all other properties of a quantum particle, angular momentum is quantized, meaning it can only equal certain discrete values, which correspond to different rotational energy states. When a particle loses angular momentum, it is said to have transitioned to a lower rotational energy state. Likewise, when a particle gains angular momentum, a positive rotational transition is said to have occurred.
In quantum physics, Fermi's golden rule is a formula that describes the transition rate from one energy eigenstate of a quantum system to a group of energy eigenstates in a continuum, as a result of a weak perturbation. This transition rate is effectively independent of time and is proportional to the strength of the coupling between the initial and final states of the system as well as the density of states. It is also applicable when the final state is discrete, i.e. it is not part of a continuum, if there is some decoherence in the process, like relaxation or collision of the atoms, or like noise in the perturbation, in which case the density of states is replaced by the reciprocal of the decoherence bandwidth.
In physics and chemistry, a selection rule, or transition rule, formally constrains the possible transitions of a system from one quantum state to another. Selection rules have been derived for electromagnetic transitions in molecules, in atoms, in atomic nuclei, and so on. The selection rules may differ according to the technique used to observe the transition. The selection rule also plays a role in chemical reactions, where some are formally spin-forbidden reactions, that is, reactions where the spin state changes at least once from reactants to products.
In differential geometry, the four-gradient is the four-vector analogue of the gradient from vector calculus.

This article describes the mathematics of the Standard Model of particle physics, a gauge quantum field theory containing the internal symmetries of the unitary product group SU(3) × SU(2) × U(1). The theory is commonly viewed as describing the fundamental set of particles – the leptons, quarks, gauge bosons and the Higgs boson.

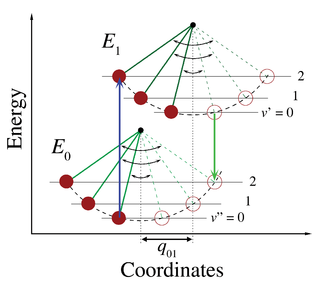
The zero-phonon line and the phonon sideband jointly constitute the line shape of individual light absorbing and emitting molecules (chromophores) embedded into a transparent solid matrix. When the host matrix contains many chromophores, each will contribute a zero-phonon line and a phonon sideband to the absorption and emission spectra. The spectra originating from a collection of identical chromophores in a matrix is said to be inhomogeneously broadened because each chromophore is surrounded by a somewhat different matrix environment which modifies the energy required for an electronic transition. In an inhomogeneous distribution of chromophores, individual zero-phonon line and phonon sideband positions are therefore shifted and overlapping.

Kasha's rule is a principle in the photochemistry of electronically excited molecules. The rule states that photon emission occurs in appreciable yield only from the lowest excited state of a given multiplicity. It is named after American spectroscopist Michael Kasha, who proposed it in 1950.

The transition dipole moment or transition moment, usually denoted for a transition between an initial state, , and a final state, , is the electric dipole moment associated with the transition between the two states. In general the transition dipole moment is a complex vector quantity that includes the phase factors associated with the two states. Its direction gives the polarization of the transition, which determines how the system will interact with an electromagnetic wave of a given polarization, while the square of the magnitude gives the strength of the interaction due to the distribution of charge within the system. The SI unit of the transition dipole moment is the Coulomb-meter (Cm); a more conveniently sized unit is the Debye (D).
The derivation of the Navier–Stokes equations as well as their application and formulation for different families of fluids, is an important exercise in fluid dynamics with applications in mechanical engineering, physics, chemistry, heat transfer, and electrical engineering. A proof explaining the properties and bounds of the equations, such as Navier–Stokes existence and smoothness, is one of the important unsolved problems in mathematics.
Resonance fluorescence is the process in which a two-level atom system interacts with the quantum electromagnetic field if the field is driven at a frequency near to the natural frequency of the atom.
In physics, relativistic quantum mechanics (RQM) is any Poincaré covariant formulation of quantum mechanics (QM). This theory is applicable to massive particles propagating at all velocities up to those comparable to the speed of light c, and can accommodate massless particles. The theory has application in high energy physics, particle physics and accelerator physics, as well as atomic physics, chemistry and condensed matter physics. Non-relativistic quantum mechanics refers to the mathematical formulation of quantum mechanics applied in the context of Galilean relativity, more specifically quantizing the equations of classical mechanics by replacing dynamical variables by operators. Relativistic quantum mechanics (RQM) is quantum mechanics applied with special relativity. Although the earlier formulations, like the Schrödinger picture and Heisenberg picture were originally formulated in a non-relativistic background, a few of them also work with special relativity.
Heat transfer physics describes the kinetics of energy storage, transport, and energy transformation by principal energy carriers: phonons, electrons, fluid particles, and photons. Heat is thermal energy stored in temperature-dependent motion of particles including electrons, atomic nuclei, individual atoms, and molecules. Heat is transferred to and from matter by the principal energy carriers. The state of energy stored within matter, or transported by the carriers, is described by a combination of classical and quantum statistical mechanics. The energy is different made (converted) among various carriers. The heat transfer processes are governed by the rates at which various related physical phenomena occur, such as the rate of particle collisions in classical mechanics. These various states and kinetics determine the heat transfer, i.e., the net rate of energy storage or transport. Governing these process from the atomic level to macroscale are the laws of thermodynamics, including conservation of energy.
Giant oscillator strength is inherent in excitons that are weakly bound to impurities or defects in crystals.


![Figure 5. Figure 1 in Edward Condon's first publication on what is now the Franck-Condon principle [Condon 1926]. Condon chose to superimpose the potential curves to illustrate the method of estimating vibrational transitions. Condon fig1 1926b.png](http://upload.wikimedia.org/wikipedia/commons/thumb/3/35/Condon_fig1_1926b.png/400px-Condon_fig1_1926b.png)

















