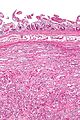
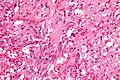
A paraganglioma is a rare neuroendocrine neoplasm that may develop at various body sites. When the same type of tumor is found in the adrenal gland, they are referred to as a pheochromocytoma. They are rare tumors, with an overall estimated incidence of 1 in 300,000. There is no test that determines benign from malignant tumors; long-term follow-up is therefore recommended for all individuals with paraganglioma.

Hemangioendotheliomas are a family of vascular neoplasms of intermediate malignancy.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.

Angiomyolipomas are the most common benign tumour of the kidney. Although regarded as benign, angiomyolipomas may grow such that kidney function is impaired or the blood vessels may dilate and burst, leading to bleeding.

Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung, and the rest of the body.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first definitively characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a cluster of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Less commonly, cases are reported in the pelvis, vulva, penis, and spine.
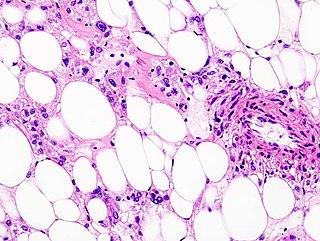
Perivascular epithelioid cell tumour, also known as PEComa or PEC tumour, is a family of mesenchymal tumours consisting of perivascular epithelioid cells (PECs). These are rare tumours that can occur in any part of the human body.

Giant-cell carcinoma of the lung (GCCL) is a rare histological form of large-cell lung carcinoma, a subtype of undifferentiated lung cancer, traditionally classified within the non-small-cell lung carcinomas (NSCLC).

DOTA-TATE is an eight amino acid long peptide, with a covalently bonded DOTA bifunctional chelator.

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.

Epithelial-myoepithelial carcinoma (EMCa) is a rare malignant tumour that typically arises in a salivary gland and consists of both an epithelial and myoepithelial component. They are predominantly found in the parotid gland and represent approximately 1% of salivary gland tumours.

Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of the mesodermal cells that form the connective tissues which support virtually all of the organs and tissues of the body. IMT was formerly termed inflammatory pseudotumor. Currently, however, inflammatory pseudotumor designates a large and heterogeneous group of soft tissue tumors that includes inflammatory myofibroblastic tumor, plasma cell granuloma, xanthomatous pseudotumor, solitary mast cell granuloma, inflammatory fibrosarcoma, pseudosarcomatous myofibroblastic proliferation, myofibroblastoma, inflammatory myofibrohistiocytic proliferation, and other tumors that develop from connective tissue cells. Inflammatory pseudotumour is a generic term applied to various neoplastic and non-neoplastic tissue lesions which share a common microscopic appearance consisting of spindle cells and a prominent presence of the white blood cells that populate chronic or, less commonly, acute inflamed tissues.

Mammary-type myofibroblastoma (MFB), also named mammary and extramammary myofibroblastoma, was first termed myofibrolastoma of the breast, or, more simply, either mammary myofibroblastoma (MMFB) or just myofibroblastoma. The change in this terminology occurred because the initial 1987 study and many subsequent studies found this tumor only in breast tissue. However, a 2001 study followed by numerous reports found tumors with the microscopic histopathology and other key features of mammary MFB in a wide range of organs and tissues. Further complicating the issue, early studies on MFB classified it as one of various types of spindle cell tumors that, except for MFB, were ill-defined. These other tumors, which have often been named interchangeably in different reports, are: myelofibroblastoma, benign spindle cell tumor, fibroma, spindle cell lipoma, myogenic stromal tumor, and solitary stromal tumor. Finally, studies suggest that spindle cell lipoma and cellular angiofibroma are variants of MFB. Here, the latter two tumors are tentatively classified as MFB variants but otherwise MFB is described as it is more strictly defined in most recent publications. The World Health Organization in 2020 classified mammary type myofibroblastoma tumors and myofibroblastoma tumors as separate tumor forms within the category of fibroblastic and myofibroblastic tumors.

An inflammatory fibroid polyp(IFP) is an uncommon digestive system tumor. J. Vanek initially identified it as a separate pathological entity in 1949 when he reported six case reports of eosinophilic infiltration in gastric submucosal granulomas. It is a single, non-encapsulated polypoid lesion that is typically submucosal. It is characterized by a large number of small blood vessels, oedematous connective tissue, and an inflammatory eosinophilic infiltrate.

Hyalinizing clear cell carcinoma (HCCC) is a rare malignant salivary gland tumour, with a good prognosis, that is usually found on the tongue or palate.
Gastroblastoma is a rare cancer that occurs in the stomach. Only six cases have been reported to date (2017).

Proliferative fasciitis and proliferative myositis (PF/PM) are rare benign soft tissue lesions that increase in size over several weeks and often regress over the ensuing 1–3 months. The lesions in PF/PM are typically obvious tumors or swellings. Historically, many studies had grouped the two descriptive forms of PF/PM as similar disorders with the exception that proliferative fasciitis occurs in subcutaneous tissues while proliferative myositis occurs in muscle tissues. In 2020, the World Health Organization agreed with this view and defined these lesions as virtually identical disorders termed proliferative fasciitis/proliferative myositis or proliferative fasciitis and proliferative myositis. The Organization also classified them as one of the various forms of the fibroblastic and myofibroblastic tumors.
Myxofibrosarcoma (MFS), although a rare type of tumor, is one of the most common soft tissue sarcomas, i.e. cancerous tumors, that develop in the soft tissues of elderly individuals. Initially considered to be a type of histiocytoma termed fibrous histiocytoma or myxoid variant of malignant fibrous histiocytoma, Angervall et al. termed this tumor myxofibrosarcoma in 1977. In 2020, the World Health Organization reclassified MFS as a separate and distinct tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Cellular angiofibroma (CAF) is a rare, benign tumor of superficial soft tissues that was first described by M. R. Nucci et al. in 1997. These tumors occur predominantly in the distal parts of the female and male reproductive systems, i.e. in the vulva-vaginal and inguinal-scrotal areas, respectively, or, less commonly, in various other superficial soft tissue areas throughout the body. CAF tumors develop exclusively in adults who typically are more than 30 years old.
Low-grade myofibroblastic sarcoma (LGMS) is a subtype of the malignant sarcomas. As it is currently recognized, LGMS was first described as a rare, atypical myofibroblastic tumor by Mentzel et al. in 1998. Myofibroblastic sarcomas had been divided into low-grade myofibroblastic sarcomas, intermediate‐grade myofibroblasic sarcomas, i.e. IGMS, and high‐grade myofibroblasic sarcomas, i.e. HGMS based on their microscopic morphological, immunophenotypic, and malignancy features. LGMS and IGMS are now classified together by the World Health Organization (WHO), 2020, in the category of intermediate fibroblastic and myofibroblastic tumors. WHO, 2020, classifies HGMS as a soft tissue tumor in the category of tumors of uncertain differentiation. This article follows the WHO classification: here, LGMS includes IGMS but not HGMS which is a more aggressive and metastasizing tumor than LGMS and consists of cells of uncertain origin.