The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). Animals that use this cycle, mainly amphibians and mammals, are called ureotelic.

Arginine is the amino acid with the formula (H2N)(HN)CN(H)(CH2)3CH(NH2)CO2H. The molecule features a guanidino group appended to a standard amino acid framework. At physiological pH, the carboxylic acid is deprotonated (−CO2−) and both the amino and guanidino groups are protonated, resulting in a cation. Only the l-arginine (symbol Arg or R) enantiomer is found naturally. Arg residues are common components of proteins. It is encoded by the codons CGU, CGC, CGA, CGG, AGA, and AGG. The guanidine group in arginine is the precursor for the biosynthesis of nitric oxide. Like all amino acids, it is a white, water-soluble solid.
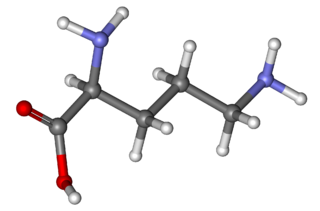
Ornithine is a non-proteinogenic α-amino acid that plays a role in the urea cycle. Ornithine is abnormally accumulated in the body in ornithine transcarbamylase deficiency. The radical is ornithyl.

The organic compound citrulline is an α-amino acid. Its name is derived from citrullus, the Latin word for watermelon. Although named and described by gastroenterologists since the late 19th century, it was first isolated from watermelon in 1914 by Japanese researchers Yatarō Koga (古賀彌太郎) and Ryō Ōtake (大嶽了) and further codified by Mitsunori Wada of Tokyo Imperial University in 1930. It has the formula H2NC(O)NH(CH2)3CH(NH2)CO2H. It is a key intermediate in the urea cycle, the pathway by which mammals excrete ammonia by converting it into urea. Citrulline is also produced as a byproduct of the enzymatic production of nitric oxide from the amino acid arginine, catalyzed by nitric oxide synthase.

In molecular biology, post-translational modification (PTM) is the covalent process of changing proteins following protein biosynthesis. PTMs may involve enzymes or occur spontaneously. Proteins are created by ribosomes, which translate mRNA into polypeptide chains, which may then change to form the mature protein product. PTMs are important components in cell signalling, as for example when prohormones are converted to hormones.

Ornithine transcarbamylase (OTC) is an enzyme that catalyzes the reaction between carbamoyl phosphate (CP) and ornithine (Orn) to form citrulline (Cit) and phosphate (Pi). There are two classes of OTC: anabolic and catabolic. This article focuses on anabolic OTC. Anabolic OTC facilitates the sixth step in the biosynthesis of the amino acid arginine in prokaryotes. In contrast, mammalian OTC plays an essential role in the urea cycle, the purpose of which is to capture toxic ammonia and transform it into urea, a less toxic nitrogen source, for excretion.

Carbamoyl phosphate is an anion of biochemical significance. In land-dwelling animals, it is an intermediary metabolite in nitrogen disposal through the urea cycle and the synthesis of pyrimidines. Its enzymatic counterpart, carbamoyl phosphate synthetase I, interacts with a class of molecules called sirtuins, NAD dependent protein deacetylases, and ATP to form carbamoyl phosphate. CP then enters the urea cycle in which it reacts with ornithine to form citrulline.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.

Ornithine transcarbamylase deficiency also known as OTC deficiency is the most common urea cycle disorder in humans. Ornithine transcarbamylase, the defective enzyme in this disorder, is the final enzyme in the proximal portion of the urea cycle, responsible for converting carbamoyl phosphate and ornithine into citrulline. OTC deficiency is inherited in an X-linked recessive manner, meaning males are more commonly affected than females.

Lysinuric protein intolerance (LPI) is an autosomal recessive metabolic disorder affecting amino acid transport. It is characterised by the body's inability to properly digest and use certain proteins. This condition leads to a buildup of lysine, arginine, and ornithine in the body, which can cause a variety of health issues. This condition leads to various metabolic complications and is typically diagnosed in infancy or early childhood.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

The enzyme argininosuccinate lyase (EC 4.3.2.1, ASL, argininosuccinase; systematic name 2-(N ω-L-arginino)succinate arginine-lyase (fumarate-forming)) catalyzes the reversible breakdown of argininosuccinate:

Citrullination or deimination is the conversion of the amino acid arginine in a protein into the amino acid citrulline. Citrulline is not one of the 20 standard amino acids encoded by DNA in the genetic code. Instead, it is the result of a post-translational modification. Citrullination is distinct from the formation of the free amino acid citrulline as part of the urea cycle or as a byproduct of enzymes of the nitric oxide synthase family.
Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.
Delta-1-pyrroline-5-carboxylate synthetase (P5CS) is an enzyme that in humans is encoded by the ALDH18A1 gene. This gene is a member of the aldehyde dehydrogenase family and encodes a bifunctional ATP- and NADPH-dependent mitochondrial enzyme with both gamma-glutamyl kinase and gamma-glutamyl phosphate reductase activities. The encoded protein catalyzes the reduction of glutamate to delta1-pyrroline-5-carboxylate, a critical step in the de novo biosynthesis of proline, ornithine and arginine. Mutations in this gene lead to hyperammonemia, hypoornithinemia, hypocitrullinemia, hypoargininemia and hypoprolinemia and may be associated with neurodegeneration, cataracts and connective tissue diseases. Alternatively spliced transcript variants, encoding different isoforms, have been described for this gene. As reported by Bruno Reversade and colleagues, ALDH18A1 deficiency or dominant-negative mutations in P5CS in humans causes a progeroid disease known as De Barsy Syndrome.

Anti-citrullinated protein antibodies (ACPAs) are autoantibodies that are directed against peptides and proteins that are citrullinated. They are present in the majority of patients with rheumatoid arthritis. Clinically, cyclic citrullinated peptides (CCP) are frequently used to detect these antibodies in patient serum or plasma.
Detection of autoantibodies against mutated citrullinated vimentin is part of rheumatoid arthritis (RA) diagnostics, especially in sera negative for rheumatoid factor. Anti-MCV antibodies are a member of the ACPA family, a group of the so-called antibodies to citrullinated protein/peptide antigens.
Solute carrier family 25, member 29 is a protein that in humans is encoded by the SLC25A29 gene. The gene is also known as CACL and C14orf69. SLC25A29 belongs to a protein family of solute carriers called the mitochondrial carriers.
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.

Amino acid score, in combination with protein digestibility, is the method used to determine if a protein is complete.


