Related Research Articles

Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.

The neonatal heel prick is a blood collection procedure done on newborns. It consists of making a pinprick puncture in one heel of the newborn to collect their blood. This technique is used frequently as the main way to collect blood from neonates. Other techniques include venous or arterial needle sticks, cord blood sampling, or umbilical line collection. This technique is often utilized for the Guthrie test, where it is used to soak the blood into pre-printed collection cards known as Guthrie cards.
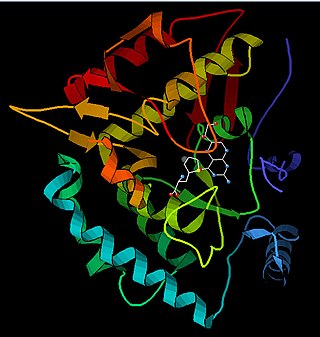
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.
Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.

Otto Neumann Knoph Sverdrup was a Norwegian sailor and Arctic explorer.

The following outline is provided as an overview of and topical guide to biochemistry:

Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.

6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors, monoamine oxidase inhibitors, and tetrahydrobiopterin. Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).
Robert Guthrie, MD, Ph.D. was an American microbiologist, best known for developing the bacterial inhibition assay used to screen infants for phenylketonuria at birth, before the development of irreversible neurological damage. Guthrie also pioneered the collection of whole blood on specially designed filter paper, commonly known as "Guthrie cards" as a sample medium that could be easily collected, transported and tested. Although Guthrie is best known for developing the test for phenylketonuria, he worked tirelessly to raise awareness of the need to screen for treatable conditions and adapted his method to early screening tests for galactosemia and maple syrup urine disease.

The enzyme phenylalanine racemase is the enzyme that acts on amino acids and derivatives. It activates both the L & D stereo isomers of phenylalanine to form L-phenylalanyl adenylate and D-phenylalanyl adenylate, which are bound to the enzyme. These bound compounds are then transferred to the thiol group of the enzyme followed by conversion of its configuration, the D-isomer being the more favorable configuration of the two, with a 7 to 3 ratio between the two isomers. The racemisation reaction of phenylalanine is coupled with the highly favorable hydrolysis of adenosine triphosphate (ATP) to adenosine monophosphate (AMP) and pyrophosphate (PP), thermodynamically allowing it to proceed. This reaction is then drawn forward by further hydrolyzing PP to inorganic phosphate (Pi), via Le Chatelier's principle.

Odd Nansen was a Norwegian architect, writer, and humanitarian. He is credited with being a co-founder of UNICEF and for his humanitarian efforts on behalf of Jews in the early years of World War II.
The Nansen Academy – Norwegian Humanistic Academy is a folk high school in Lillehammer, Norway.
Events in the year 1973 in Norway.
Hyperphenylalaninemia is a medical condition characterized by mildly or strongly elevated concentrations of the amino acid phenylalanine in the blood. Phenylketonuria (PKU) can result in severe hyperphenylalaninemia. Phenylalanine concentrations are routinely screened in newborns by the neonatal heel prick, which takes a few drops of blood from the heel of the infant. Standard phenylalanine concentrations in unaffected persons are about 2-6mg/dl phenylalanine concentrations in those with untreated hyperphenylalaninemia can be up to 20 mg/dL. Measurable IQ deficits are often detected as phenylalanine levels approach 10 mg/dL. Phenylketonuria (PKU)-like symptoms, including more pronounced developmental defects, skin irritation, and vomiting, may appear when phenylalanine levels are near 20 mg/dL .Hyperphenylalaninemia is a recessive hereditary metabolic disorder that is caused by the body's failure to convert phenylalanine to tyrosine as a result of the entire or partial absence of the enzyme phenylalanine hydroxylase.

Polhøgda is the home of the Fridtjof Nansen Institute. It was originally built as the private home of Norwegian explorer Fridtjof Nansen. The manor home's architecture is Roman Revival, and the former estate lies between Lysaker and Fornebu in Bærum, Norway.

Oluf Christian Dietrichson was a Norwegian explorer and military officer. He was a member of the Greenland expedition of 1888 led by Fridtjof Nansen, the first documented crossing of Greenland.

Norwegian of the Century was a poll carried out by the Norwegian Broadcasting Corporation in 2005, the 100-year anniversary of Norwegian independence. The poll was SMS-based and over 400,000 Norwegians voted over the course of the year. To qualify as "Norwegian of the Century", the nominee must have lived between 1905 and 2005. All Norwegians were eligible for nomination, and there were initially 600 people on the list. A "Great Norwegian Committee" consisting of Nils Arne Eggen, Astrid Nøklebye Heiberg, Guri Hjeltnes, Harald Norvik, Erling Sandmo and Cathrine Sandnes narrowed the list down to 50. Another poll was conducted, again SMS-based, with the results presented live on NRK1 on 17 December 2005. The winner, with 41% of the vote, was King Olav V. Former Prime Minister Einar Gerhardsen was second with 24%, followed by Erik Bye with 15%. The results for the top 50 spots were as follows:
Harvey Louis Levy is an American biochemical geneticist, pediatrician, physician scientist and academic. He is Senior Physician in Medicine and Genetics at Boston Children’s Hospital and Professor of Pediatrics at Harvard Medical School.

Louis Isaac Woolf was a British biochemist who played a crucial role in the early detection and the treatment of phenylketonuria (PKU).
References
- ↑ Ole Daniel Enersen. "Følling's disease or Følling's syndrome". whonamedit.com. Retrieved 1 February 2018.
- ↑ Fölling, Asbjörn (1934). "Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität". Hoppe-Seyler's Zeitschrift für physiologische Chemie. 227 (1–4): 169–181. doi:10.1515/bchm2.1934.227.1-4.169.
- ↑ Centerwall, S. A.; Centerwall, W. R. (2000). "The discovery of phenylketonuria: the story of a young couple, two affected children, and a scientist". Pediatrics. 105 (1 Pt 1): 89–103. doi:10.1542/peds.105.1.89. PMID 10617710. S2CID 35922780.
- ↑ Svein Atle Skålevåg. "Asbjørn Følling". Store norske leksikon. Retrieved 1 February 2018.
- ↑ Sverre O. Lie. "Asbjørn Følling". Norsk biografisk leksikon. Retrieved 1 February 2018.
- ↑ Jason Gonzalez; Monte S. Willis. "Ivar Asbjörn Følling: Discovered Phenylketonuria (PKU)". Laboratory Medicine, Volume 41, Issue 2, 1 February 2010, Pages 118–119. Retrieved 1 February 2018.
- ↑ "Dr. Ivar Asbjørn Følling – The Man Who Discovered PKU Disorder". pkutest.com. 12 July 2012. Retrieved 1 February 2018.
- ↑ "1949 A. Følling - Den matematisk-naturvitenskapelige klasse". Fridtjof Nansens Belønning for Fremragende Forskning. 12 July 2012. Archived from the original on 20 February 2009. Retrieved 1 February 2018.
- ↑ "Anders Jahres medisinske priser - 1960 professor Asbjørn Følling, Oslo". University of Oslo. Retrieved 1 February 2018.
- ↑ "President John F. Kennedy with Joseph P. Kennedy, Jr., Foundation Awards Recipients". John F. Kennedy Presidential Library And Museum. Retrieved 1 February 2018.
- ↑ Jon Gunnar Arntzen. "Gunnerus-medaljen". Store norske leksikon. Retrieved 1 February 2018.