As organelles, lipid droplets function as a storage compartment for a cell's metabolic energy reserves. Lipid droplets are the only cellular compartment dedicated to the storage of TAGs and other neutral lipids, making these organelles crucial for both energy storage functions and for the aversion of cellular lipotoxicity. Lipid droplets also serve as a reservoir for cholesterol esters (CEs) and fat-soluble vitamins, as well as many other polymeric lipids.[1]
Both the appearance and the distribution of lipid droplets changes by cell type, and may reflect the specialized functions of a given type of cell. Generally, the diameter of lipid droplets ranges from 0.1-5µm in non-adipocyte cells, but increases to over 100µm in white adipocytes.[2] Research on lipid droplet function has proved crucial in both health and disease, as these organelles are known to support many large-scale biological processes such as development and aging.
The role of lipid droplets outside of neutral lipid storage remains a topic of ongoing research.
Significance
Everyday, cells within the human body rely on the metabolic energy stores found in lipid droplets to survive. Sufficient levels of energy reserves are found and kept in specialized cells called adipocytes (or fat cells), which are crucial for human survival. Unlike other cells, adipocytes are specialized for storage of metabolic energy reserves, and as such, an abundance of lipid droplets are typically found within them. During times of starvation, these lipid droplet reserves decrease within adipocytes and are scarcely found. However, sustained caloric excess (over-eating) stimulates the growth of these lipid droplet reserves as they accumulate excess lipids from caloric surplus. Caloric excess stimulates not only the expansion of lipid droplets, but also the expansion of adipocytes (or fat cells), in a process known as adipose hypertrophy.
Notably, lipid droplets (LDs) bear a unique structure relative to all other cellular organelles. LDs emerge from the endoplasmic reticulum, where many remain continuous within cytoplasmic leaflets of the endoplasmic reticulum phospholipid bilayer itself.[3] LDs thus bear a phospholipid monolayer, which envelopes a highly dynamic core of hydrophobic neutral lipids. While all LDs are known to share this structure, the behavior and morphologies of these organelles are both diverse and extremely dynamic.[3]
Lipid droplets (red) and nuclei (blue) are shown in adipocytes from adult D. melanogaster, stained with Oil Red O and DAPI, respectively.
Lipid droplets (orange), the cell membrane (blue), and nuclei (cyan) are shown in adipocytes from adult D. melanogaster.
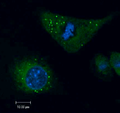
Lipid droplets (green) and nuclei (blue) are shown in adipocytes from adult D. melanogaster, stained with BODIPY 505/510 and DAPI, respectively.
Outer Membrane Monolayer
Unlike other organelles, the outer membrane of lipid droplets (LDs) is composed of a phospholipid monolayer. LD formation begins within the phospholipid bilayer of the endoplasmic reticulum (ER), where a cytoplasmic leaflet of the phospholipid bilayer "buds" as neutral lipids accumulate at its center (core). This phospholipid monolayer ultimately becomes the LD droplet surface, and remains continuous with the phospholipid bilayer of ER.[3] Hydrophobic neutral lipids enveloped by the phospholipid monolayer remain highly dynamic at its core, where they are stored or hydrolyzed in accordance with cellular energetic needs. Throughout further LD maturation, the outer phospholipid monolayer of the organelle is thought to remain connected to the endoplasmic reticulum via a hairpin-like "stalk" formation between the two organelles.[3]
Membrane Surface Proteins
The surface of the LD monolayer is decorated with a vast and diverse repertoire of proteins, the number of which varies from species to species. In yeast, approximately 40 different LD proteins have been cataloged using proteomics-based approaches, while the mammalian LD proteome is known to consist of over 100 proteins to date.[3][4][5][6][7] LD surface proteins are known to regulate and dictate several aspects of the LD life-cycle, including LD budding, growth, turnover, and interaction with other cellular organelles, such as mitochondria.[3][8][9] Inherently, the LD proteome is highly dynamic and represents a key area of interest in modern lipid research.
The first and best-characterized family of lipid droplet associated proteins is the perilipin protein family, consisting of five proteins. These include perilipin 1 (PLIN1), perilipin 2 (PLIN2), perilipin 3 (PLIN3), perilipin 4 (PLIN4) and perilipin 5 (PLIN5).[10][11]Proteomics studies have elucidated the association of many other families of proteins to the lipid surface, including those involved in membrane trafficking, vesicle docking, endocytosis and exocytosis.[12]
Lipid Droplet Core
At their core, lipid droplets (LDs) contain a highly dynamic, hydrophobic deposit of neutral lipids, such as triacylglycerols (TAGs) and cholesteryl esters (CEs). In most cells, metabolic energy is stored in the form of fatty acids (FAs), which are the building blocks of triacylglycerols (TAGs) or fat. Lipid droplets are the only cellular compartment dedicated to the storage of TAGs and other neutral lipids, making these organelles crucial for both energy storage functions and the aversion of lipotoxicity. In adipocytes (or fat cells), TAGs are the predominant component of the LD core. However, in other cell types, various ratios of TAGs and CEs are found in the LD core.
Demarcation between TAGs and CEs within the LD core has not been observed by conventional electron microscopy (EM) alone, although segregation within the core may exist in certain circumstances.[2] Some evidence exists to suggest partitioning among lipid esters in the LD core, such as concentric lipid ester layers observed by cryoelectron microscopy,[2][13] and island-like fracture faces seen by freeze-fracture electron microscopy.[2][13][14] Membrane-like structures have also been observed in the LD core in more specialized cell types.[2][15][16]
Generally, LD heterogeneity refers to observable differences in LD size, abundance, distribution, location, core lipid composition, or proteome composition about the organelle. Discrete combinations of these features are thought to shape general functional differences between LD populations, many of which may be present across different cell types, as well as within the same cell.[3] Of all factors used to characterize specific LD types, the surface proteome has proven most useful. Features associated with a specific LD type are largely determined by the proteins on the LD surface, many of which facilitate LD growth or shrinkage such as lipid enzymes, as well as scaffolding proteins and factors that mediate interactions with other organelles.[3][4][5][6][7] LD heterogeneity is best characterized within cells of a given cell type, where it may thus reflect relative changes in cellular metabolic state or become indicative of physiological disease. However, LD heterogeneity within a single cell likely reflects the discrete functions of different LD subpopulations in metabolic homeostasis, in both health and disease.
Differences Across Cell Types
Canonically, the core function of the LD as a lipid storage depot is conserved across cell types and species. However, cellular identity (and thus specialization) determines the threshold for LD utility and thus caps LD heterogeneity by cell type.
Lipid Droplet Biogenesis
Lipid droplet (LD) biogenesis begins at the membrane of the endoplasmic reticulum (ER). Despite over a decade of modern research, the process of LD formation has yet to be fully understood.
LD biogenesis is triggered by the accumulation of neutral lipids within the membrane of the endoplasmic reticulum,[3][18][19][20][21] which occurs in response to elevated dietary carbohydrate or lipid intake. In simplest terms, lipid droplets form should the rate of neutral lipid synthesis at the ER exceed the ER membrane's capacity to accommodate those lipids, causing them to phase-separate and bud into lipid droplets.[3][22] To date, LD assembly appears to follow a single robust mechanism, regardless of the type of neutral lipid involved.[3] Nevertheless, LD biogenesis has been extensively characterized for triacylglycerols (TAGs). While the formation of lipid droplets remains dependent on the availability of free fatty acids and other metabolites, the general process can be nevertheless summarized by the following sections.
Availability of Fatty Acids
A number of metabolic reactions must first occur within the cell in order for synthesis of the main lipid droplet (LD) component triacylglycerol (TAG) to occur. A more detailed explanation of fatty acid synthesis is available here, but the topic (in full) remains outside the scope of this article. Nevertheless, several reactions relevant to TAG synthesis are summarized below:
In brief, TAG and phospholipids are generated from glucose-derived glycerol and mitochondrial-derived fatty acids.[22]Acetyl-CoA is the precursor used for fatty acid synthesis in the cytosol;[22] however, acetyl-CoA is unable to be shuttled directly into the cytosol in its original form. Originally, acetyl-CoA is generated in the mitochondria from pyruvate molecules derived from glucose via glycolysis.[23][22] Within the mitochondria, acetyl-CoA typically combines with oxaloacetate and serves as a substrate for the synthesis of citrate as part of the well known citric acid cycle. Notably, the inner mitochondrial membrane is impermeable to acetyl-CoA, and as such, a specialized shuttle system must be used to import acetyl-CoA into the cytosol for fatty acid production.[22] This process, known as the citrate–malate shuttle, relies on the tricarboxylate transport protein to import citrate into the cytosol, where it is then split into acetyl-CoA and oxaloacetate by the enzyme ATP citrate lyase (ACL).[22] Cytosolic acetyl-CoA is then available for use in fatty acid and cholesterol synthesis, but oxaloacetate must be reduced to malate in order to reenter the mitochondria. Malate dehydrogenase reduces cytosolic oxaloacetate by coupling NADH oxidation to NAD+,[22] and malate produced by this reaction can be transported back into the mitochondria, thus completing the namesake of the citrate–malate shuttle.
Reaction 1. Formation of Malonyl-CoA
In the first reaction of fatty acid synthesis, acetyl-CoA is irreversibly carboxylated by acetyl-CoA carboxylase (ACC) to form malonyl-CoA. The formation of malonyl-CoA is the rate-limiting step of fatty acid synthesis.
This step provides the malonyl-CoA substrate for use in fatty acid synthesis.
Reaction 2. Formation of Acetyl-ACP and Malonyl-ACP
In the second reaction of fatty acid synthesis, acetyl transacylase and malonyl transacylase catalyze the formation of acetyl-ACP and malonyl-ACP, respectively. The transacylase enzymes use the sulfhydryl group of ACP to release CoA and form acetyl-ACP and malonyl-ACP.
This step activates acetyl-CoA and malonyl-CoA for use in fatty acid synthesis.
Reaction 3. Condensation of Acetyl-ACP and Malonyl-ACP
During the third step of fatty acid synthesis, acetyl-ACP and malonyl-ACP undergo a condensation reaction catalyzed by β-ketoacyl-ACP synthase, which produces a four-carbon acetoacetyl-ACP molecule and one molecule of CO2. The condensation reaction is shown in (c).
This is the first condensation reaction of fatty acid synthesis.
Reaction 4. Reduction of Acetoacetyl-ACP to β-hydroxyacyl-ACP
During the fourth reaction of fatty acid synthesis, acetoacetyl-ACP is reduced by 3-ketoacyl-ACP reductase to form one molecule of β-hydroxyacyl-ACP. The reaction scheme for the reduction is shown in (d). NADPH is used as the reducing agent.
This is the first reduction reaction of fatty acid synthesis.
Reaction 5. Dehydration of β-hydroxyacyl-ACP to Enoyl-ACP
During the fifth reaction of fatty acid synthesis, β-hydroxyacyl-ACP undergoes a dehydration reaction catalyzed by 3-hydroxyacyl-ACP dehydratase. The reaction scheme for the dehydration is shown in (e). One molecule of water is removed from a β-hydroxyacyl intermediate to form a double bond, saturating the chain and producing enoyl-ACP.
This is the first dehydration reaction of fatty acid synthesis.
Reaction 6. Reduction of Enoyl-ACP to Butyryl-ACP
During the sixth reaction of fatty acid synthesis, enoyl-ACP is reduced by enoyl-ACP reductase to form butyryl-ACP. The reaction scheme is shown in (f). Herein, the double bond of the trans-2-enoyl-ACP molecule is reduced to a saturated acyl-ACP using NADPH as the reducing agent.
This is the second reduction reaction of fatty acid synthesis.
Fatty Acid Synthesis
Fatty acid synthesis begins in the cytosol. During the first reaction, irreversible carboxylation of acetyl-CoA to malonyl-CoA is catalyzed by the biotin-dependent enzyme acetyl-CoA carboxylase (ACC).[22] Notably, the conversion of acetyl-CoA to malonyl-CoA is the rate-limiting step of fatty acid synthesis. Acetyl-CoA carboxylase (ACC) thus represents the rate-limiting enzyme in fatty acid synthesis; ACC activity is stimulated by increasing concentrations of cytosolic citrate, and inhibited by increasing concentrations of the fatty acid palmitate.[22]
After malonyl-CoA becomes available by virtue of ACC, fatty acid synthase (FAS) is then able to complete a series of reactions to form the 16-carbon molecule palmitate. FAS is a complex, multifunctional protein containing seven different catalytic sites: acetyl transacylase, malonyl transacylase, β-ketoacyl synthase, β-ketoacyl carrier protein (ACP) reductase, 3-hydroxyacyl-ACP dehydratase, enoyl-ACP reductase, and thioesterase.[22] These different enzymes are covalently linked within the FAS complex, allowing for intermediates to be handled efficiently from one active site to another without leaving the assembly.[22] After the completion of the first reaction by ACC, fatty acid synthesis thus continues on the FAS complex.
During the second reaction of fatty acid synthesis, acetyl transacylase and malonyl transacylase catalyze the formation of acetyl-ACP and malonyl-ACP, respectively.[22] Acetyl transacylase transfers the acetyl group of acetyl-CoA onto the sulfhydryl group of Acyl Carrier Protein (ACP), releasing CoA and forming acetyl-ACP.[22] An equivalent reaction occurs for malonyl-CoA, in which malonyl transacylase transfers the malonyl group from malonyl-CoA to the sulfhydryl group of Acyl Carrier Protein (ACP), releasing CoA and forming malonyl-ACP.[22] These two reactions are essential, as they prime the acetyl and malonyl groups for condensation in the subsequent chain elongation reaction step.[22]
After the production of acetyl-ACP and malonyl-ACP, fatty acid synthesis begins to cycle through repetitions of the following reaction sequence: condensation → reduction → dehydration → reduction.[22] Ultimately, this elongation reaction sequence repeats through 7 cycles to form one molecule of (16C) palmitate, as malonyl-CoA (the carbon donor) adds 2 carbons to the growing chain per cycle.
During the third reaction of fatty acid synthesis, acetyl-ACP and malonyl-ACP undergo a condensation reaction catalyzed by the enzyme β-ketoacyl-ACP synthase (also known as acyl-malonyl-ACP condensing enzyme), which produces the four-carbon acetoacetyl-ACP molecule and one molecule of CO2.[22] Notably, the reaction of two-carbon acetyl-ACP with three-carbon malonyl-ACP is more favorable than that of two, two-carbon acetyl-ACP molecules reacting together.[22]
The fourth step of fatty acid elongation is the reduction of acetoacyl-ACP to β-hydroxyacyl-ACP, in a reaction catalyzed by3-ketoacyl-ACP reductase.[22] Herein, the electron donorNADPH is used as the reducing agent, ultimately converting the β-keto group of β-ketoacyl-ACP into the β-hydroxyl group of β-hydroxyacyl-ACP.[22]
The fifth step of fatty acid elongation is the dehydration of β-hydroxyacyl-ACP to enoyl-ACP, in a reaction catalyzed by3-hydroxyacyl-ACP dehydratase.[22] 3-hydroxyacyl-ACP dehydratase removes one molecule of H2O to form a double bond between the C2–C3 carbons of β-hydroxyacyl-ACP, thereby saturating the chain and producing enoyl-ACP.[22]
The sixth step of fatty acid elongation is the reduction of enoyl-ACP to butyryl-ACP, in a reaction catalyzed byenoyl-ACP reductase.[22] Herein, enoyl-ACP reductase reduces the C2–C3 double bond of enoyl-ACP into a saturated acyl-ACP using one molecule NADPH as the electron donor.[22] The production of butyryl-ACP thus marks the completion of the first cycle of fatty acid elongation, and the reaction sequence thereafter repeats again (condensation → reduction → dehydration → reduction).[22]
At the beginning of the second cycle, butyryl-ACP condenses with a molecule of malonyl-ACP, forming the six-carbon β-ketoacyl-ACP molecule and one molecule of CO2.[22] The next three reactions within the second cycle (reduction → dehydration → reduction) convert the six-carbon β-ketoacyl-ACP into a six-carbon ACP molecule, which thus marks the completion of the second cycle of fatty acid elongation, and a third cycle can thereafter begin.[22] These elongation cycles continue (x7) until a (16C) acyl-ACP molecule is formed. Thereafter, the (16C) acyl-ACP is hydrolyzed by a thioesterase to form one molecule of palmitate and one molecule of ACP.[22]
Palmitate produced by FAS can be used in the generation of even longer fatty acids, in a process unsurprisingly catalyzed by elongase enzymes, which lengthen palmitate to yield long chain fatty acids.[22] Alternatively, palmitate can undergo desaturation reactions, in a process catalyzed by desaturase enzymes, which ultimately generate unsaturated fatty acids.[22] Further elongation of palmitate requires the addition of a CoA thioester to the molecule in an ATP-dependent reaction, which is catalyzed by acyl-CoA synthetase.[22] Further elongation occurs through the subsequent additions of malonyl-CoA molecules onto palmitate, or onto other saturated or unsaturated fatty acyl-CoA substrates.[22] These further elongation reactions are catalyzed by fatty acyl synthase enzyme, which is located on the cytosolic face of the endoplasmic reticulum (ER).[22] Herein, these condensation reactions are driven by the decarboxylation of the additional malonyl-CoA substrates.[22] Unlike the former elongation cycles, which produced the sixteen-carbon palmitate substrate, the further elongation of palmitate does not involve ACP and does not rely on any multifunctional enzyme (ie., FAS).[22]
Synthesis of Triacylglycerol (TAG)
Once fatty acids become available, they can be used to generate triacylglycerol (TAG) through their addition to glycerol 3-phosphate. Again, a small number of metabolic reactions must occur within the cell prior to TAG production, beginning with the formation of glycerol 3-phosphate, and followed by the formation of multiple fatty acid intermediates. First, the glycolytic intermediate dihydroxyacetone (DHA) is converted into glycerol 3-phosphate, in a reaction catalyzed by the enzyme glycerol 3-phosphate dehydrogenase.[22] Once made, glycerol 3-phosphate is primed for three subsequent additions of fatty acid chains, which will ultimately produce one molecule of TAG.
Lens formation is known to occur at critical concentrations ranging from 5 to 10mol%, though bilayer properties may modulate these ranges. Factors such as phospholipid head group composition, saturation and length of phospholipidacyl chains, and membrane surface tension and curvature are all known to influence these critical concentrations.[3][25] Nevertheless, the lens structure remains embedded within the ER bilayer, where the cytoplasmic leaflet of the ER bilayer continues to envelop the growing lipid droplet core, ultimately becoming the phospholipid monolayer of the nascent LD itself.[3][24][25] Sustained TAG synthesis causes smaller existing lenses to converge, producing larger lenses which begin to bud away from the ER membrane and outwards towards the cytosol.
Although lipid lens formation can be driven solely through increasing concentrations of neutral lipids, the process remains tightly regulated by residential ER-membrane proteins. Of all proteins known to mediate LD assembly, the evolutionarily conserved ER-membrane protein Seipin has gained considerable research attention over the last ten years.[24][26][27][28][29]Seipin, a homo-oligomeric integral membrane protein, marks sites of LD biogenesis, and each cytoplasmic LD appears to associate with at least one Seipin focus.[24][21][30]Cryoelectron microscopy studies have demonstrated Seipin protomers localizing at sites of LD nucleation, where the luminal domains of up to 12 protomers organize into a homoligomeric ring structure.[3][28][31][32][33]Loss-of-function studies have revealed that this ring-like assembly is crucial for Seipin function.[3][32][33] Within the Seipin luminal domain, a hydrophobic helix (HH) projects from the luminal ring into the ER bilayer, and has been found to concentrate TAGs within the ring itself, thereby catalyzing the formation of neutral lipid lenses.[3] Interestingly, HH projections from the Seipin ring complex are capable of catalyzed LD formation, even when TAG concentrations within the ER bilayer were too low to produce phase separation.[3]
Study of early LD nucleation is greatly complicated by the transient nature of early intermediates in vitro. Thus, characterization of LD nucleation has largely relied on techniques applied in silico and modern molecular dynamics simulations.[3][34] Conventional electron microscopytomography experiments provided the first in vivo evidence for the lens model of LD biogenesis, where lens-like structures of approximately 50nm in diameter were observed in yeast, following the induction of TAG biosynthesis.[3][35]
Lipid Droplet Budding
As the lipid droplet (LD) continues to grow and bud away from the endoplasmic reticulum (ER) bilayer, it remains attached through a small membrane stalk or hairpin-like structure.[3][18][20] Both the directionality and the efficacy of the budding process are unsurprisingly influenced by various properties of the ER bilayer itself, including its lipid composition, curvature, and surface tension.[3] Directionality itself is imposed by localized asymmetry between the two leaflets of the ER bilayer, a process thought to derive from differences in leaflet protein and/or phospholipid composition at sites of LD formation.[3][36][37] Differences between the leaflets reduce the surface tension of the cytosolic-facing monolayer, thereby facilitating budding in that direction.[3] Given that LD budding seems to nearly always favor formation towards the cytosol, factors controlling localized asymmetry between the leaflets must be tightly regulated; however, the precise mechanisms controlling this process remain ill-defined.[3]
Of the proteins known to facilitate LD budding, the ER-residential membrane protein fat-inducing transcript 2 (FIT2) has been subject to considerable research attention over the last ten years.[3][38][39][40][35][41] FIT proteins, also known as fat storage-inducing transmembrane (FITM) proteins, are an evolutionarily conserved family of proteins known for their namesake promotion of lipid storage.[42] The role of these proteins in facilitating LD formation proves crucial, as loss of FIT proteins halts LD budding, thereby inducing an accumulation of neutral lipid lenses within the ER bilayer.[3][35] Yet, mechanistic details on FIT proteins remain ill-defined.[3] Several recent studies suggest roles for these proteins in regulating ER phospholipid composition and membrane architecture,[3][38][39] while others suggest FIT2 controls the directionality of LD budding by regulating DAG levels at sites of LD biogenesis.[3][40]In vitro murine studies have revealed that loss of FIT2 produces global deficits in ER homeostasis, with several defects in ER organization, bilayer phospholipid composition, and TAG metabolism having been reported.[3][38][39] To date, it remains unclear whether FIT2 shapes LD biogenesis solely via global regulation of ER homeostasis, or through an additional role in facilitating LD budding and maturation.[3]
As directionality itself is driven by localized asymmetry between two leaflets of the ER bilayer, differences in leaflet protein and/or phospholipid compositions at sites of LD formation may pose as key mechanistic components of LD budding.[3] Tension imbalances within the bilayer can be generated via asymmetric addition of phospholipids and/or proteins about the LD formation site, through either the recruitment of essential machinery for LD biogenesis, or through crowding imposed by neighboring ER-bound activities.[3] The perilipin (PLIN) family of proteins, which are by far the most abundant proteins associated with the LD surface, are thought to utilize this mechanism to facilitate LD budding.[3] Notably, PLIN3 is known to lower monolayer tension via insertion of an amphipathic helix into the cytosolic leaflet of the ER membrane, thereby facilitating LD budding.[3][37][36] Recruitment of PLIN3 to sites of LD biogenesis is stimulated by the production of TAG precursors, such as phosphatidic acid and diacylglycerols (DAGs), suggesting a complex link between TAG synthesis and factors regulating ER bilayer tension.[3][43][44]
Lipid Droplet Growth & Maturation
Further expansion of the nascent lipid droplet (LD) relies on the continuous phospholipid supply from the ER to the LD monolayer via the membrane stalk, which maintains the connection of the two organelles.[3] Following budding from the ER bilayer, nascent lipid droplets (LDs) continue to grow through expansion of the triacylglycerol (TAG) core. Further expansion relies on the continuous phospholipid supply from the ER to the LD monolayer via the membrane stalk, which maintains the connection of the two organelles.[3] Interestingly, TAG can be supplied via synthesis in the ER and direct transfer through the membrane stalk, or via direct TAG synthesis on the LD surface.[3] TAG synthesis on the LD surface requires relocalization of several enzymes from the ER onto the LD monolayer, including the acyltransferase DGAT2.[3][45] During periods of rapid LD expansion, synthesis of new phospholipids is necessary to maintain phospholipid homeostasis about the LD monolayer, which requires relocalization of several enzymes involved in this process to the LD surface.[3][4][46]
Critically, the ER-residential protein seipin ensures the fidelity of LD growth and maturation via stabilization of the membrane stalk that connects the LD to the ER bilayer.[3][28] During LD biogenesis, seipin protomers assemble into a decameric, cage-like structure at sites of LD assembly, forming a stable ring of luminal domains at the cage floor with transmembrane domains at the cage sides and top.[28] These transmembrane segments interact with adjacent protomers in two distinct, alternating conformations, which are produced via changes in switch regions located between the luminal and transmembrane domains.[28] Structural studies propose a model for LD formation in which the closed conformer of the seipin cage enables TAG phase separation during neutral lipid lens formation, whereas the open conformer enables for LD budding and expansion.[28] Loss of seipin during LD maturation is known to induce regression of TAG back into the ER, thus highlighting the importance of seipin-stabilization at the membrane stalk during LD biogenesis and beyond.[3][30]
In higher eukaryotes, a fraction of LDs are thought to eventually detach from the ER bilayer.[3] However, the reasons for this and the mechanism of ER detachment are still unknown.[3] Interestingly, in vitro studies in insect and mammalian cells suggest that LD detachment from the ER is reversible.[3][47] Reattachment appears to utilize components of the COPI coatomer complex, which is usually known for its roles in retrograde Golgi-to-ER trafficking.[3][47][48]
Protein Regulation of Lipid Droplet Formation
Though LD biogenesis can be viewed as a biophysical process driven purely by thermodynamic principles, individual stages of LD formation within cells are highly regulated by several protein machineries.[3] Lipid lens formation can be driven solely through increasing concentrations of neutral lipids, yet the process remains tightly regulated by homo-oligomers of the conserved ER-residential membrane protein seipin and its interacting partners.[24][49]Seipin marks sites of LD biogenesis, and each cytoplasmic LD appears to associate with at least one seipin focus.[24][21][30] Recently, the results of several structural studies suggest a role for seipin in the promotion of TAG aggregation and facilitation of neutral lipid lens formation at defined sites about the ER bilayer.[24][26][27][28][29]Loss of seipin is not sufficient for ablation of LD biogenesis, but rather results in aberrant LD biogenesis and produces organelles with abnormal morphology, protein composition, and functions.[24][6][19][50] Several studies have observed seipin preventing spontaneous coalescence of lenses at random sites throughout the ER bilayer,[24][27][26] but its role in modulating these processes remains incompletely understood. Mutations in the BSCL2 gene, which encodes seipin, have been reported in patients with hereditary forms of lipodystrophy, providing the first link between the protein and lipid storage processes.[3][51]
Other interacting proteins such as lipid droplet assembly factor 1 (LDAF1) may also modulate seipin's ability to bind to TAG.[3][52][26] LDAF1 reportedly binds seipin, forming an ~600 kDa oligomeric complex that copurifies with TAG.[52] During LD biogenesis, LDAF1-seipin complexes have been observed at sites of lens formation, where re-localization of LDAF1 to the ER bilayer recruits seipin and promotes LD formation at these sites.[52] As LDs coalesce within the bilayer and begin to bud away from the ER, LDAF1 dissociates from seipin and moves onto the LD surface.[52] Interestingly, loss of LDAF1 produces similar albeit milder aberrations to LD biogenesis, in comparison to loss of seipin.[3][52][26] To date, the available data support a model in which the formation of neutral lipid lenses catalyzed by seipin begins to expand at sites marked by the seipin/LDAF1 complex.[3] The long-chain-fatty-acid-CoA ligase 3 (ACSL3) enzyme also localizes to these early sites of LD formation, and its catalytic activity may contribute in part to the rapid synthesis of lipids that fuels further lens growth.[3][20]
Protein Targeting to Lipid Droplets
Mechanisms by which the large and highly dynamic set of proteins traffic to the lipid droplet (LD) surface have been an area of intense modern research. Unlike proteins targeting other cellular organelles, LD proteins have no distinct targeting signal or localization sequence embedded within their amino acid sequence.[3] Protein association with the surface monolayer occurs via hydrophobic hairpins, lipidated domains, and amphipathic helices, or by binding to another protein on the LD surface.[3] LD proteins are thus classified into two main categories (class I and class II) by virtue of their pathway for targeting the LD surface: the ERTOLD pathway or the CYTOLD pathway.
Class I Proteins: ERTOLD Pathway
Class I proteins enter the LD surface by virtue of the ERTOLD pathway, where they are first inserted into the ER membrane and are thereafter trafficked onto the LD surface. Class I proteins form characteristic stable membrane association with the phospholipid monolayer of the LD, with this most commonly achieved via insertion of hydrophobic hairpin segments into the membrane and core.[3] Within this configuration, class I proteins retain cytosolic exposure of both the C-terminus and N-terminus, thus creating ample opportunity for other cytosolic proteins to act upon the LD in cell signaling cascades.[3] Class I proteins explicitly lack luminal domains, as these would preclude their movement within the ER bilayer and onto the LD surface.[3][53] As such, polytopic membrane proteins can never be class I proteins, nor will they enter the LD surface via the ERTOLD pathway.[3][53]
Although the structural features of class I proteins allow for their free movement between the ER bilayer and the LD surface, it remains unclear how the relative partitioning between the two organelles is controlled.[3] Targeting to LDs is favored when the hairpin structure of class I proteins is flanked by positively charged residues, which may facilitate more energetically stable conformations on the LD surface compared to the ER bilayer.[3][54][55] Certain class I proteins display an affinity for triacylglycerol (TAG) about their hairpin loop, which drives their concentration at the LD surface.[3][56] Conversely, some class I proteins may be actively retained on the ER surface through protein-protein interactions.[3] For example, UBXD8 is selectively partitioned onto the ER bilayer when bound to UBAC2, a polytopic ER membrane protein.[3][57][58] Selective degradation of certain hairpin-containing proteins in the ER results in their effective accumulation at the LD surface.[3][4][59][60] Although these examples and putative mechanisms offer insights as to the regulation of class I protein partitioning between the ER and LD surface, a complete mechanistic understanding of this process has yet to be uncovered.
Several imaging studies have revealed that some class I proteins localize to LDs during the earliest stages of LD biogenesis, while other proteins target LDs much later.[3][53][18] Such observations suggest the existence of at least two ERTOLD pathways that effectively stage LD protein targeting in an orderly fashion.[3] Interestingly, recent research reports seem to further evidence the existence of such "early" and "late" ERTOLD pathways. Recently, some class I proteins have been shown to target LDs during the budding phase of early LD biogenesis, where they appeared to diffuse onto LDs from the ER through the Seipin-stabilized neck.[3][61] By contrast, other class I proteins were shown to be initially rejected at the Seipin gate and appeared to follow another distinct route, which depends on machinery typically associated with anterograde vesicular traffic in the early secretory pathway.[3][61]
Factors relevant to this latent ERTOLD pathway include components of the COPII coat complex (Sec23 and Sec24), ER exit site factors (Sec12, Sec16, Sar1, and Tango1), membrane tethering complexes (TRAPP and COG complex subunits), and Rab proteins as well as SNARE proteins known for their importance in membrane fusion.[3] These findings led to the idea that the late ERTOLD targeting pathway may involve the establishment of distinct membrane bridges between the ER and LD, which may function in a Seipin-independent fashion.[3] In the absence of Seipin, late ERTOLD pathway cargo appears much earlier upon the LD surface,[61] further supporting earlier studies in yeast and mammalian cells, which showed the central role of Seipin was not only concentrating triacyl glycerol (TAG), but also regulating the LD proteome by gating movement of class I proteins between the ER and LD surface.[3][6][19]
Class II Proteins: CYTOLD Pathway
Class II proteins enter the LD surface by virtue of the CYTOLD pathway, where they are recruited directly from the cytosol to the phospholipid monolayer, supported in some cases by chaperone proteins.[3] Class II proteins associate with LDs via a variety of mechanisms, including direct binding onto other LD proteins or association with the lipid monolayer using a lipid anchor.[3][62] Most frequently, class II proteins are found bound to LDs via amphipathic alpha-helices (AHs), in which hydrophobic and polarresidues partition to opposite sides of the helix.[3][37][63][64][65][66]
Interestingly, the specificity of AH-containing proteins for the LD monolayer appears to be caused by phospholipid packing defects and the higher surface tension of the LD monolayer relative to the phospholipid bilayer of all other cellular organelles.[3][65] This specificity is further increased in AH-containing proteins whose AHs are known to interact directly with TAG.[64][67] However, not all AH-containing LD proteins behave as class II proteins.[3] Recently, it was shown that some class I protein trafficking to the LD monolayer is also mediated by AHs.[3][68] Comparison of multiple AHs suggests that class I behavior is favored for AHs with a reduced number of charged residues along the polar face, possibly due to the greater allowance for extensive interactions with the phospholipid side chains.[3][68] Conversely, class II behavior is favored for AHs with a greater presence of charged residues, as such reduces the axial hydrophobic surface of the AH.[68]
An example is the endocannabinoids that are unsaturated fatty acid derivatives, which mainly are considered to be synthesised "on demand" from phospholipid precursors residing in the cell membrane, but may also be synthesised and stored in intracellular lipid droplets and released from those stores under appropriate conditions.[70]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.