The following is a list of software packages and applications for biocybernetics research.
Contents
- Data formats and specifications
- Libraries for software development
- Software products
- See also
- Other collections
- References
The following is a list of software packages and applications for biocybernetics research.

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is often referred to as computational biology, though the distinction between the two terms is often disputed.

Computational biology refers to the use of data analysis, mathematical modeling and computational simulations to understand biological systems and relationships. An intersection of computer science, biology, and big data, the field also has foundations in applied mathematics, chemistry, and genetics. It differs from biological computing, a subfield of computer science and engineering which uses bioengineering to build computers.
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.
The Open Bioinformatics Foundation is a non-profit, volunteer-run organization focused on supporting open source programming in bioinformatics. The mission of the foundation is to support the development of open source toolkits for bioinformatics, organise developer-centric hackathon events and generally assist in the development and promotion of open source software development in the life sciences. The foundation also organises and runs the annual Bioinformatics Open Source Conference, a satellite meeting of the Intelligent Systems for Molecular Biology conference. The foundation participates in the Google Summer of Code, acting as an umbrella organisation for individual bioinformatics-related projects.
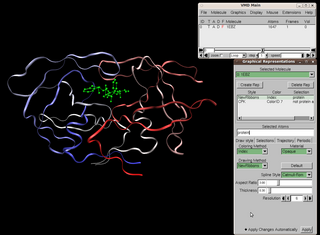
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
The Systems Biology Markup Language (SBML) is a representation format, based on XML, for communicating and storing computational models of biological processes. It is a free and open standard with widespread software support and a community of users and developers. SBML can represent many different classes of biological phenomena, including metabolic networks, cell signaling pathways, regulatory networks, infectious diseases, and many others. It has been proposed as a standard for representing computational models in systems biology today.

The Chemistry Development Kit (CDK) is computer software, a library in the programming language Java, for chemoinformatics and bioinformatics. It is available for Windows, Linux, Unix, and macOS. It is free and open-source software distributed under the GNU Lesser General Public License (LGPL) 2.0.
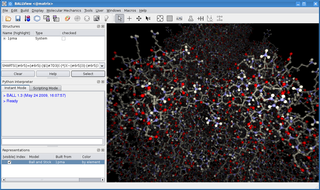
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
BioModels is a free and open-source repository for storing, exchanging and retrieving quantitative models of biological interest created in 2006. All the models in the curated section of BioModels Database have been described in peer-reviewed scientific literature.
The Systems Biology Ontology (SBO) is a set of controlled, relational vocabularies of terms commonly used in systems biology, and in particular in computational modeling.
Systems immunology is a research field under systems biology that uses mathematical approaches and computational methods to examine the interactions within cellular and molecular networks of the immune system. The immune system has been thoroughly analyzed as regards to its components and function by using a "reductionist" approach, but its overall function can't be easily predicted by studying the characteristics of its isolated components because they strongly rely on the interactions among these numerous constituents. It focuses on in silico experiments rather than in vivo.
Physiomics is a systematic study of physiome in biology. Physiomics employs bioinformatics to construct networks of physiological features that are associated with genes, proteins and their networks. A few of the methods for determining individual relationships between the DNA sequence and physiological function include metabolic pathway engineering and RNAi analysis. The relationships derived from methods such as these are organized and processed computationally to form distinct networks. Computer models use these experimentally determined networks to develop further predictions of gene function.
COPASI is an open-source software application for creating and solving mathematical models of biological processes such as metabolic networks, cell-signaling pathways, regulatory networks, infectious diseases, and many others.
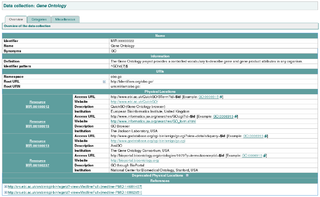
The MIRIAM Registry, a by-product of the MIRIAM Guidelines, is a database of namespaces and associated information that is used in the creation of uniform resource identifiers. It contains the set of community-approved namespaces for databases and resources serving, primarily, the biological sciences domain. These shared namespaces, when combined with 'data collection' identifiers, can be used to create globally unique identifiers for knowledge held in data repositories. For more information on the use of URIs to annotate models, see the specification of SBML Level 2 Version 2.
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.
libRoadRunner is a C/C++ software library that supports simulation of SBML based models.. It uses LLVM to generate extremely high-performance code and is the fastest SBML-based simulator currently available. Its main purpose is for use as a reusable library that can be hosted by other applications, particularly on large compute clusters for doing parameter optimization where performance is critical. It also has a set of Python bindings that allow it to be easily used from Python as well as a set of bindings for Julia.
Smoldyn is an open-source software application for cell-scale biochemical simulations. It uses particle-based simulation, meaning that it simulates each molecule of interest individually, in order to capture natural stochasticity and yield nanometer-scale spatial resolution. Simulated molecules diffuse, react, are confined by surfaces, and bind to membranes in similar manners as in real biochemical systems.