| |
| Names | |
|---|---|
| Other names Oxodiperoxymolybdenum(pyridine)(hexamethylphosphoric triamide) [1] Vedejs Reagent | |
| Identifiers | |
3D model (JSmol) | |
| ChemSpider | |
PubChem CID |
|
CompTox Dashboard (EPA) | |
| |
| |
| Properties | |
| C11H23MoN4O6P | |
| Molar mass | 434.25 g·mol−1 |
| Appearance | Yellow crystals [1] |
| Melting point | 103–105 °C (217–221 °F; 376–378 K) (dec) [1] |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
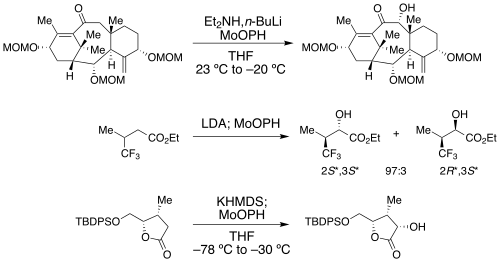
MoOPH, also known as oxodiperoxymolybdenum(pyridine)-(hexamethylphosphoric triamide), is a reagent used in organic synthesis. [1] It contains a molybdenum(VI) center with multiple oxygen ligands, coordinated with pyridine and HMPA ligands, although the HMPA can be replaced by DMPU. [2] It is an electrophilic source of oxygen that reacts with enolates and related structures, and thus can be used for alpha-hydroxylation of carbonyl-containing compounds. [3] Other reagents used for alpha-hydroxylation via enol or enolate structures include Davis oxaziridine, oxygen, and various peroxyacids (see Rubottom oxidation). This reagent was first utilized by Edwin Vedejs as an efficient alpha-hydroxylating agent in 1974 and an effective preparative procedure was later published in 1978. [4]