
Serum protein electrophoresis is a laboratory test that examines specific proteins in the blood called globulins. The most common indications for a serum protein electrophoresis test are to diagnose or monitor multiple myeloma, a monoclonal gammopathy of uncertain significance (MGUS), or further investigate a discrepancy between a low albumin and a relatively high total protein. Unexplained bone pain, anemia, proteinuria, chronic kidney disease, and hypercalcemia are also signs of multiple myeloma, and indications for SPE. Blood must first be collected, usually into an airtight vial or syringe. Electrophoresis is a laboratory technique in which the blood serum is applied to either an acetate membrane soaked in a liquid buffer, or to a buffered agarose gel matrix, or into liquid in a capillary tube, and exposed to an electric current to separate the serum protein components into five major fractions by size and electrical charge: serum albumin, alpha-1 globulins, alpha-2 globulins, beta 1 and 2 globulins, and gamma globulins.

IgA nephropathy (IgAN), also known as Berger's disease, or synpharyngitic glomerulonephritis, is a disease of the kidney and the immune system; specifically it is a form of glomerulonephritis or an inflammation of the glomeruli of the kidney. Aggressive Berger's disease can attack other major organs, such as the liver, skin and heart.
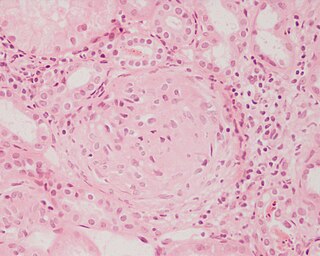
Glomerulonephritis (GN) is a term used to refer to several kidney diseases. Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name, but not all diseases necessarily have an inflammatory component.

Cryoglobulinemia is a medical condition in which the blood contains large amounts of pathological cold sensitive antibodies called cryoglobulins – proteins that become insoluble at reduced temperatures. This should be contrasted with cold agglutinins, which cause agglutination of red blood cells.

Membranous glomerulonephritis (MGN) is a slowly progressive disease of the kidney affecting mostly people between ages of 30 and 50 years, usually white people.
Monoclonal gammopathy of undetermined significance (MGUS) is a plasma cell dyscrasia in which plasma cells or other types of antibody-producing cells secrete a myeloma protein, i.e. an abnormal antibody, into the blood; this abnormal protein is usually found during standard laboratory blood or urine tests. MGUS resembles multiple myeloma and similar diseases, but the levels of antibodies are lower, the number of plasma cells in the bone marrow is lower, and it rarely has symptoms or major problems. However, since MGUS can lead to multiple myeloma, which develops at the rate of about 1.5% a year, or other symptomatic conditions, yearly monitoring is recommended.

Monoclonal gammopathy, also known as paraproteinemia, is the presence of excessive amounts of myeloma protein or monoclonal gamma globulin in the blood. It is usually due to an underlying immunoproliferative disorder or hematologic neoplasms, especially multiple myeloma. It is sometimes considered equivalent to plasma cell dyscrasia. The most common form of the disease is monoclonal gammopathy of undetermined significance.

Rapidly progressive glomerulonephritis (RPGN) is a syndrome of the kidney that is characterized by a rapid loss of kidney function, with glomerular crescent formation seen in at least 50% or 75% of glomeruli seen on kidney biopsies. If left untreated, it rapidly progresses into acute kidney failure and death within months. In 50% of cases, RPGN is associated with an underlying disease such as Goodpasture syndrome, systemic lupus erythematosus or granulomatosis with polyangiitis; the remaining cases are idiopathic. Regardless of the underlying cause, RPGN involves severe injury to the kidneys' glomeruli, with many of the glomeruli containing characteristic glomerular crescents.

Membranoproliferative glomerulonephritis (MPGN) is a type of glomerulonephritis caused by deposits in the kidney glomerular mesangium and basement membrane (GBM) thickening, activating the complement system and damaging the glomeruli.
A myeloma protein is an abnormal antibody (immunoglobulin) or a fragment thereof, such as an immunoglobulin light chain, that is produced in excess by an abnormal monoclonal proliferation of plasma cells, typically in multiple myeloma or Monoclonal gammopathy of undetermined significance. Other terms for such a protein are monoclonal protein, M protein, M component, M spike, spike protein, or paraprotein. This proliferation of the myeloma protein has several deleterious effects on the body, including impaired immune function, abnormally high blood viscosity, and kidney damage.
In hematology, plasma cell dyscrasias are a spectrum of progressively more severe monoclonal gammopathies in which a clone or multiple clones of pre-malignant or malignant plasma cells over-produce and secrete into the blood stream a myeloma protein, i.e. an abnormal monoclonal antibody or portion thereof. The exception to this rule is the disorder termed non-secretory multiple myeloma; this disorder is a form of plasma cell dyscrasia in which no myeloma protein is detected in serum or urine of individuals who have clear evidence of an increase in clonal bone marrow plasma cells and/or evidence of clonal plasma cell-mediated tissue injury. Here, a clone of plasma cells refers to group of plasma cells that are abnormal in that they have an identical genetic identity and therefore are descendants of a single genetically distinct ancestor cell.

Mesangial proliferative glomerulonephritis (MesPGN) is a morphological pattern characterized by a numerical increase in mesangial cells and expansion of the extracellular matrix within the mesangium of the glomerulus. The increase in the number of mesangial cells can be diffuse or local and immunoglobulin and/or complement deposition can also occur. MesPGN is associated with a variety of disease processes affecting the glomerulus, though can be idiopathic. The clinical presentation of MesPGN usually consists of hematuria or nephrotic syndrome. Treatment is often consistent with the histologic pattern of and/or disease process contributing to mesangial proliferative glomerulonephritis, and usually involves some form of immunosuppressant.
Diffuse proliferative glomerulonephritis (DPGN) is a type of glomerulonephritis that is the most serious form of renal lesions in SLE and is also the most common, occurring in 35% to 60% of patients. In absence of SLE, DPGN pathology looks more like Membranoproliferative glomerulonephritis
Free light chains (FLCs) are immunoglobulin light chains that are found in the serum (blood) in an unbound (free) state. In recent decades, measuring the amount of free light chains (FLCs) in the blood has become a practical clinical test. FLC tests can be used to diagnose and monitor diseases like multiple myeloma and amyloidosis.
Complement factor H-related protein 5 (CFHR5) nephropathy is a form of inherited kidney disease which is endemic in Cyprus and is caused by a mutation in the gene CFHR5. It is thought to affect up to 1:6000 Cypriots but has not been reported in anybody who is not of Cypriot descent.

Light chain deposition disease (LCDD) is a rare blood cell disease which is characterized by deposition of fragments of infection-fighting immunoglobulins, called light chains (LCs), in the body. LCs are normally cleared by the kidneys, but in LCDD, these light chain deposits damage organs and cause disease. The kidneys are almost always affected and this often leads to kidney failure. About half of people with light chain deposition disease also have a plasma cell dyscrasia, a spectrum of diseases that includes multiple myeloma, Waldenström's macroglobulinemia, and the monoclonal gammopathy of undetermined significance premalignant stages of these two diseases. Unlike in AL amyloidosis, in which light chains are laid down in characteristic amyloid deposits, in LCDD, light chains are deposited in non-amyloid granules.
Onconephrology is a specialty in nephrology that deals with the study of kidney diseases in cancer patients. A nephrologist who takes care of patients with cancer and kidney disease is called an onconephrologist. This branch of nephrology encompasses nephrotoxicity associated with existing and novel chemotherapeutics, kidney disease as it pertains to stem cell transplant, paraneoplastic kidney disorders, paraproteinemias, electrolyte disorders associated with cancer, and more as discussed below.

LECT2 Amyloidosis (ALECT2) is a form of amyloidosis caused by the LECT2 protein. It was found to be the third most common cause of amyloidosis in a set of more than 4,000 individuals studied at the Mayo Clinic; the first and second most common forms the disorder were AL amyloidosis and AA amyloidosis, respectively. Amyloidosis is a disorder in which the abnormal deposition of a protein in organs and/or tissues gradually leads to organ failure and/or tissue injury.
Monoclonal immunoglobulin deposition disease, or MIDD, is a disease characterised by the deposition of monoclonal immunoglobulins on the basement membrane of the kidney. Monoclonal immunoglobulins are produced by monoclonal plasma cells, which are found in a variety of plasma cell dyscrasias. The deposition of monoclonal immunoglobulins on the basement membrane of the kidney causes renal impairment. As well as the kidney, MIDD may also affect the liver, heart, peripheral nerves, lung and skin.
Crystal-storing histiocytosis is a form of histiocytosis which mostly occurs in people with monoclonal gammopathies. Histiocytosis is an excessive number of histiocytes. In the vast majority of crystal-storing histiocytosis cases, immunoglobulins accumulate within the cytoplasm of histiocytes; in rare cases clofazimine, cystine, silica, or Charcot–Leyden crystals may be found in the histiocytes instead. Non-immunoglobulin crystal-storing histiocytosis is mostly associated with non-malignant disorders, such as chronic inflammation or autoimmune abnormality conditions such as rheumatoid arthritis, Crohn's disease, or Helicobacter pylori gastritis. It may be a localised or generalised disease. Examples of locations where histiocytosis may occur include the lungs, pleura, stomach, kidney, bone marrow, thyroid, thymus, and parotid gland. The disease is described as generalised if two or more unrelated sites are involved.