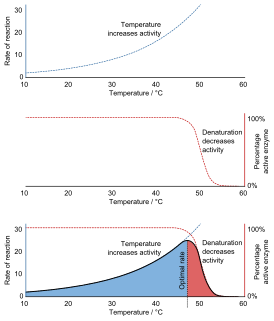
Denaturation is a process in which proteins or nucleic acids lose the quaternary structure, tertiary structure, and secondary structure which is present in their native state, by application of some external stress or compound such as a strong acid or base, a concentrated inorganic salt, an organic solvent, radiation or heat. If proteins in a living cell are denatured, this results in disruption of cell activity and possibly cell death. Protein denaturation is also a consequence of cell death. Denatured proteins can exhibit a wide range of characteristics, from conformational change and loss of solubility to aggregation due to the exposure of hydrophobic groups. Denatured proteins lose their 3D structure and therefore cannot function.
Grammar theory to model symbol strings originated from work in computational linguistics aiming to understand the structure of natural languages. Probabilistic context free grammars (PCFGs) have been applied in probabilistic modeling of RNA structures almost 40 years after they were introduced in computational linguistics.
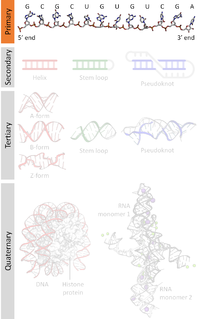
A nucleic acid sequence is a succession of bases signified by a series of a set of five different letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
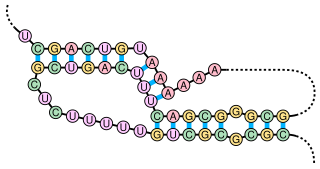
A pseudoknot is a nucleic acid secondary structure containing at least two stem-loop structures in which half of one stem is intercalated between the two halves of another stem. The pseudoknot was first recognized in the turnip yellow mosaic virus in 1982. Pseudoknots fold into knot-shaped three-dimensional conformations but are not true topological knots.

Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
Nucleic acid thermodynamics is the study of how temperature affects the nucleic acid structure of double-stranded DNA (dsDNA). The melting temperature (Tm) is defined as the temperature at which half of the DNA strands are in the random coil or single-stranded (ssDNA) state. Tm depends on the length of the DNA molecule and its specific nucleotide sequence. DNA, when in a state where its two strands are dissociated, is referred to as having been denatured by the high temperature.
Ribosomal frameshifting, also known as translational frameshifting or translational recoding, is a biological phenomenon that occurs during translation that results in the production of multiple, unique proteins from a single mRNA. The process can be programmed by the nucleotide sequence of the mRNA and is sometimes affected by the secondary, 3-dimensional mRNA structure. It has been described mainly in viruses, retrotransposons and bacterial insertion elements, and also in some cellular genes.
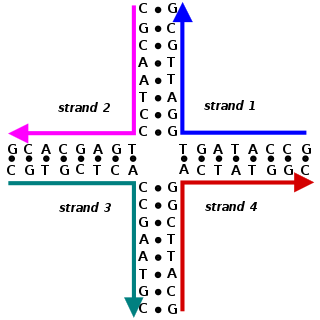
Nucleic acid design is the process of generating a set of nucleic acid base sequences that will associate into a desired conformation. Nucleic acid design is central to the fields of DNA nanotechnology and DNA computing. It is necessary because there are many possible sequences of nucleic acid strands that will fold into a given secondary structure, but many of these sequences will have undesired additional interactions which must be avoided. In addition, there are many tertiary structure considerations which affect the choice of a secondary structure for a given design.
taveRNA is a software suite for RNA/DNA secondary structure. It is developed in the laboratories for computational biology of the School of Computing Science at the Simon Fraser University. The suite is composed by alteRNA, for RNA density fold computing, inteRNA, for RNA-RNA interaction prediction, piRNA, for predicting the joint partition function, equilibrium concentration, ensemble energy, and melting temperature for two RNA sequences, pRuNA, a sequence based pruning RNA interaction search engine, and smyRNA, a platform independent C program novel ab initio ncRNA finder.

Nucleic acid tertiary structure is the three-dimensional shape of a nucleic acid polymer. RNA and DNA molecules are capable of diverse functions ranging from molecular recognition to catalysis. Such functions require a precise three-dimensional tertiary structure. While such structures are diverse and seemingly complex, they are composed of recurring, easily recognizable tertiary structure motifs that serve as molecular building blocks. Some of the most common motifs for RNA and DNA tertiary structure are described below, but this information is based on a limited number of solved structures. Many more tertiary structural motifs will be revealed as new RNA and DNA molecules are structurally characterized.

Nucleic acid structure refers to the structure of nucleic acids such as DNA and RNA. Chemically speaking, DNA and RNA are very similar. Nucleic acid structure is often divided into four different levels: primary, secondary, tertiary, and quaternary.
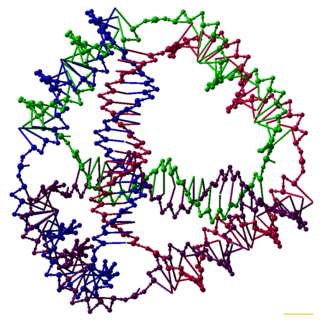
DNA nanotechnology is the design and manufacture of artificial nucleic acid structures for technological uses. In this field, nucleic acids are used as non-biological engineering materials for nanotechnology rather than as the carriers of genetic information in living cells. Researchers in the field have created static structures such as two- and three-dimensional crystal lattices, nanotubes, polyhedra, and arbitrary shapes, and functional devices such as molecular machines and DNA computers. The field is beginning to be used as a tool to solve basic science problems in structural biology and biophysics, including applications in X-ray crystallography and nuclear magnetic resonance spectroscopy of proteins to determine structures. Potential applications in molecular scale electronics and nanomedicine are also being investigated.

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNA's and RNA's tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.
Niles A. Pierce is an American mathematician, bioengineer, and professor at the California Institute of Technology. He is a leading researcher in the fields of molecular programming and dynamic nucleic acid nanotechnology. His research is focused on kinetically controlled DNA and RNA self-assembly. Pierce is working on applications in bioimaging.
The 3' splice site of the influenza A virus segment 7 pre-mRNA can adopt two different types of RNA structure: a pseudoknot and a hairpin. This conformational switch is proposed to play a role in RNA alternative splicing and may influence the production of M1 and M2 proteins produced by splicing of this pre-mRNA.
A neutral network is a set of genes all related by point mutations that have equivalent function or fitness. Each node represents a gene sequence and each line represents the mutation connecting two sequences. Neutral networks can be thought of as high, flat plateaus in a fitness landscape. During neutral evolution, genes can randomly move through neutral networks and traverse regions of sequence space which may have consequences for robustness and evolvability.

Robert Dirks was an American chemist known for his theoretical and experimental work in DNA nanotechnology. Born in Thailand to a Thai Chinese mother and American father, he moved to Spokane, Washington at a young age. Dirks was the first graduate student in Niles Pierce's research group at the California Institute of Technology, where his dissertation work was on algorithms and computational tools to analyze nucleic acid thermodynamics and predict their structure. He also performed experimental work developing a biochemical chain reaction to self-assemble nucleic acid devices. Dirks later worked at D. E. Shaw Research on algorithms for protein folding that could be used to design new pharmaceuticals.
The ViennaRNA Package is a set of standalone programs and libraries used for prediction and analysis of RNA secondary structures. The source code for the package is distributed freely and compiled binaries are available for Linux, macOS and Windows platforms. The original paper has been cited over 2000 times.

