





Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene. [5] [6] [7]
Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene. [5] [6] [7]
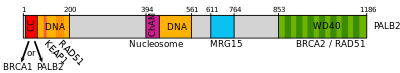

This gene encodes a protein that functions in genome maintenance (double strand break repair). This protein binds to and colocalizes with the breast cancer 2 early onset protein (BRCA2) in nuclear foci and likely permits the stable intranuclear localization and accumulation of BRCA2. [5] PALB2 binds the single strand DNA and directly interacts with the recombinase RAD51 to stimulate strand invasion, a vital step of homologous recombination, [15] PALB2 can function synergistically with a BRCA2 chimera (termed piccolo, or piBRCA2) to further promote strand invasion. [15]
Variants in the PALB2 gene are associated with an increased risk of developing breast cancer [16] of magnitude similar to that associated with BRCA2 mutations [17] and PALB2-deficient cells are sensitive to PARP inhibitors. [15]
PALB2 was recently identified as a susceptibility gene for familial pancreatic cancer by scientists at the Sol Goldman Pancreatic Cancer Research Center at Johns Hopkins. This has paved for the way for developing a new gene test for families where pancreatic cancer occurs in multiple family members. [18] Tests for PALB2 have been developed by Ambry Genetics [19] and Myriad Genetics [20] that are now available.
Prophylactic mastectomy should be considered for women that had breast cancer and a PALB2 mutation. [21] [22]
Biallelic mutations in PALB2 (also known as FANCN), similar to biallelic BRCA2 mutations, cause Fanconi anemia. [7]
Mutations in this gene have been associated with an increased risk of ovarian, breast and pancreatic cancer. [23]
PALB2 mutant male mice have reduced fertility. [24] This reduced fertility appears to be due to germ cell attrition resulting from a combination of unrepaired DNA breaks during meiosis and defective synapsis of the X and Y chromosomes. The function of homologous recombination during meiosis appears to be repair of DNA damages, particularly double-strand breaks (also see Origin and function of meiosis).[ citation needed ] The PALB2-BRCA1 interaction is likely important for repairing such damages during male meiosis.