
Enterobacteria phage λ is a bacterial virus, or bacteriophage, that infects the bacterial species Escherichia coli. It was discovered by Esther Lederberg in 1950. The wild type of this virus has a temperate life cycle that allows it to either reside within the genome of its host through lysogeny or enter into a lytic phase, during which it kills and lyses the cell to produce offspring. Lambda strains, mutated at specific sites, are unable to lysogenize cells; instead, they grow and enter the lytic cycle after superinfecting an already lysogenized cell.
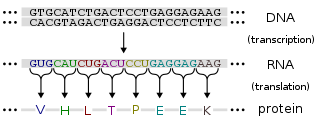
Protein production is the biotechnological process of generating a specific protein. It is typically achieved by the manipulation of gene expression in an organism such that it expresses large amounts of a recombinant gene. This includes the transcription of the recombinant DNA to messenger RNA (mRNA), the translation of mRNA into polypeptide chains, which are ultimately folded into functional proteins and may be targeted to specific subcellular or extracellular locations.

In molecular biology, RNA polymerase, or more specifically DNA-directed/dependent RNA polymerase (DdRP), is an enzyme that catalyzes the chemical reactions that synthesize RNA from a DNA template.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

A cloning vector is a small piece of DNA that can be stably maintained in an organism, and into which a foreign DNA fragment can be inserted for cloning purposes. The cloning vector may be DNA taken from a virus, the cell of a higher organism, or it may be the plasmid of a bacterium. The vector contains features that allow for the convenient insertion of a DNA fragment into the vector or its removal from the vector, for example through the presence of restriction sites. The vector and the foreign DNA may be treated with a restriction enzyme that cuts the DNA, and DNA fragments thus generated contain either blunt ends or overhangs known as sticky ends, and vector DNA and foreign DNA with compatible ends can then be joined by molecular ligation. After a DNA fragment has been cloned into a cloning vector, it may be further subcloned into another vector designed for more specific use.
DNA primase is an enzyme involved in the replication of DNA and is a type of RNA polymerase. Primase catalyzes the synthesis of a short RNA segment called a primer complementary to a ssDNA template. After this elongation, the RNA piece is removed by a 5' to 3' exonuclease and refilled with DNA.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.
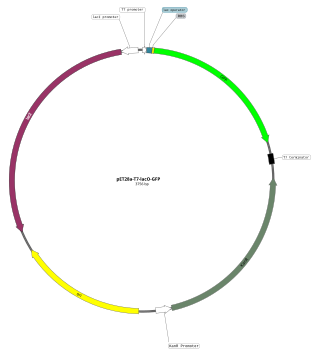
An expression vector, otherwise known as an expression construct, is usually a plasmid or virus designed for gene expression in cells. The vector is used to introduce a specific gene into a target cell, and can commandeer the cell's mechanism for protein synthesis to produce the protein encoded by the gene. Expression vectors are the basic tools in biotechnology for the production of proteins.

Bacteriophage T7 is a bacteriophage, a virus that infects bacteria. It infects most strains of Escherichia coli and relies on these hosts to propagate. Bacteriophage T7 has a lytic life cycle, meaning that it destroys the cell it infects. It also possesses several properties that make it an ideal phage for experimentation: its purification and concentration have produced consistent values in chemical analyses; it can be rendered noninfectious by exposure to UV light; and it can be used in phage display to clone RNA binding proteins.
The rpoB gene encodes the β subunit of bacterial RNA polymerase and the homologous plastid-encoded RNA polymerase (PEP). It codes for 1342 amino acids in E. coli, making it the second-largest polypeptide in the bacterial cell. It is targeted by the rifamycin family of antibacterials, such as rifampin. Mutations in rpoB that confer resistance to rifamycins do so by altering the protein's drug-binding residues, thereby reducing affinity for these antibiotics.

A termination signal is a sequence that signals the end of transcription or translation. Termination signals are found at the end of the part of the chromosome being transcribed during transcription of mRNA. Termination signals bring a stop to transcription, ensuring that only gene-encoding parts of the chromosome are transcribed. Transcription begins at the promoter when RNA polymerase, an enzyme that facilitates transcription of DNA into mRNA, binds to a promoter, unwinds the helical structure of the DNA, and uses the single-stranded DNA as a template to synthesize RNA. Once RNA polymerase reaches the termination signal, transcription is terminated. In bacteria, there are two main types of termination signals: intrinsic and factor-dependent terminators. In the context of translation, a termination signal is the stop codon on the mRNA that elicits the release of the growing peptide from the ribosome.

T7 RNA Polymerase is an RNA polymerase from the T7 bacteriophage that catalyzes the formation of RNA from DNA in the 5'→ 3' direction.
Missense mRNA is a messenger RNA bearing one or more mutated codons that yield polypeptides with an amino acid sequence different from the wild-type or naturally occurring polypeptide. Missense mRNA molecules are created when template DNA strands or the mRNA strands themselves undergo a missense mutation in which a protein coding sequence is mutated and an altered amino acid sequence is coded for.

fis is an E. coli gene encoding the Fis protein. The regulation of this gene is more complex than most other genes in the E. coli genome, as Fis is an important protein which regulates expression of other genes. It is supposed that fis is regulated by H-NS, IHF and CRP. It also regulates its own expression (autoregulation). Fis is one of the most abundant DNA binding proteins in Escherichia coli under nutrient-rich growth conditions.
In molecular cloning, a vector is any particle used as a vehicle to artificially carry a foreign nucleic sequence – usually DNA – into another cell, where it can be replicated and/or expressed. A vector containing foreign DNA is termed recombinant DNA. The four major types of vectors are plasmids, viral vectors, cosmids, and artificial chromosomes. Of these, the most commonly used vectors are plasmids. Common to all engineered vectors are an origin of replication, a multicloning site, and a selectable marker.
The lacUV5 promoter is a mutated promoter from the Escherichia coli lac operon which is used in molecular biology to drive gene expression on a plasmid. lacUV5 is very similar to the classical lac promoter, containing just 2 base pair mutations in the -10 hexamer region, compared to the lac promoter. LacUV5 is among the most commonly used promoters in molecular biology because it requires no additional activators and it drives high levels of gene expression.

cII or transcriptional activator II is a DNA-binding protein and important transcription factor in the life cycle of lambda phage. It is encoded in the lambda phage genome by the 291 base pair cII gene. cII plays a key role in determining whether the bacteriophage will incorporate its genome into its host and lie dormant (lysogeny), or replicate and kill the host (lysis).
Charles Clifton Richardson is an American biochemist and professor at Harvard University. Richardson received his undergraduate education at Duke University, where he majored in medicine. He received his M.D. at Duke Medical School in 1960. Richardson works as a professor at Harvard Medical School, and he served as editor/associate editor of the Annual Review of Biochemistry from 1972 to 2003. Richardson received the American Chemical Society Award in Biological Chemistry in 1968, as well as numerous other accolades.
The T7 expression system is used in the field of microbiology to clone recombinant DNA using strains of E. coli. It is the most popular system for expressing recombinant proteins in E. coli.
Bacteriophage AP205 is a plaque-forming bacteriophage that infects Acinetobacter bacteria. Bacteriophage AP205 is a protein-coated virus with a positive single-stranded RNA genome. It is a member of the family Fiersviridae, consisting of particles that infect Gram-negative bacteria such as E. coli.






