
An autosome is any chromosome that is not a sex chromosome. The members of an autosome pair in a diploid cell have the same morphology, unlike those in allosomal pairs, which may have different structures. The DNA in autosomes is collectively known as atDNA or auDNA.

BRCA2 and BRCA2 are human genes and their protein products, respectively. The official symbol and the official name are maintained by the HUGO Gene Nomenclature Committee. One alternative symbol, FANCD1, recognizes its association with the FANC protein complex. Orthologs, styled Brca2 and Brca2, are common in other vertebrate species. BRCA2 is a human tumor suppressor gene, found in all humans; its protein, also called by the synonym breast cancer type 2 susceptibility protein, is responsible for repairing DNA.

Succinate dehydrogenase [ubiquinone] cytochrome b small subunit, mitochondrial (CybS), also known as succinate dehydrogenase complex subunit D (SDHD), is a protein that in humans is encoded by the SDHD gene. Names previously used for SDHD were PGL and PGL1. Succinate dehydrogenase is an important enzyme in both the citric acid cycle and the electron transport chain. Hereditary PGL-PCC syndrome is caused by a parental imprint of the SDHD gene. Screening can begin by 6 years of age.
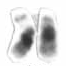
Chromosome 21 is one of the 23 pairs of chromosomes in humans. Chromosome 21 is both the smallest human autosome and chromosome, with 45 million base pairs representing about 1.5 percent of the total DNA in cells. Most people have two copies of chromosome 21, while those with three copies of chromosome 21 have Down syndrome.
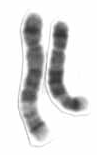
Chromosome 5 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 5 spans about 182 million base pairs and represents almost 6% of the total DNA in cells. Chromosome 5 is the 5th largest human chromosome, yet has one of the lowest gene densities. This is partially explained by numerous gene-poor regions that display a remarkable degree of non-coding and syntenic conservation with non-mammalian vertebrates, suggesting they are functionally constrained.
Adenomatous polyposis coli (APC) also known as deleted in polyposis 2.5 (DP2.5) is a protein that in humans is encoded by the APC gene. The APC protein is a negative regulator that controls beta-catenin concentrations and interacts with E-cadherin, which are involved in cell adhesion. Mutations in the APC gene may result in colorectal cancer and desmoid tumors.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

22q13 deletion syndrome, known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
A chromosomal abnormality, chromosomal anomaly, chromosomal aberration, chromosomal mutation, or chromosomal disorder is a missing, extra, or irregular portion of chromosomal DNA. These can occur in the form of numerical abnormalities, where there is an atypical number of chromosomes, or as structural abnormalities, where one or more individual chromosomes are altered. Chromosome mutation was formerly used in a strict sense to mean a change in a chromosomal segment, involving more than one gene. Chromosome anomalies usually occur when there is an error in cell division following meiosis or mitosis. Chromosome abnormalities may be detected or confirmed by comparing an individual's karyotype, or full set of chromosomes, to a typical karyotype for the species via genetic testing.

Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.

Usher syndrome type-1G protein is a protein that in humans is encoded by the USH1G gene.

Transcriptional regulator ATRX also known as ATP-dependent helicase ATRX, X-linked helicase II, or X-linked nuclear protein (XNP) is a protein that in humans is encoded by the ATRX gene.

GATA3 is a transcription factor that in humans is encoded by the GATA3 gene. Studies in animal models and humans indicate that it controls the expression of a wide range of biologically and clinically important genes.

5'-AMP-activated protein kinase subunit gamma-2 is an enzyme that in humans is encoded by the PRKAG2 gene.

DNA polymerase delta catalytic subunit(DPOD1) is an enzyme that is encoded in the human by the POLD1 gene, in the DNA polymerase delta complex. DPOD1 is responsible for synthesizing the lagging strand of DNA, and has also been implicated in some activities at the leading strand. The DPOD1 subunit encodes both DNA polymerizing and exonuclease domains, which provide the protein an important second function in proofreading to ensure replication accuracy during DNA synthesis, and in a number of types of replication-linked DNA repair following DNA damage.

Polypeptide N-acetylgalactosaminyltransferase 3 is an enzyme that in humans is encoded by the GALNT3 gene.

Centrosomal protein of 290 kDa is a protein that in humans is encoded by the CEP290 gene. CEP290 is located on the Q arm of chromosome 12.
Leucine-rich PPR motif-containing protein, mitochondrial is a protein that in humans is encoded by the LRPPRC gene. Transcripts ranging in size from 4.8 to 7.0 kb which result from alternative polyadenylation have been reported for this gene.
Cerebral dysgenesis–neuropathy–ichthyosis–keratoderma syndrome is a neurocutaneous condition caused by mutation in the SNAP29 gene.
Ataxia-pancytopenia syndrome is a rare autosomal dominant disorder characterized by cerebellar ataxia, peripheral neuropathies, pancytopenia and a predilection to myelodysplastic syndrome and acute myeloid leukemia.



