
Retinoblastoma (Rb) is a rare form of cancer that rapidly develops from the immature cells of a retina, the light-detecting tissue of the eye. It is the most common primary malignant intraocular cancer in children, especially those under 3 years old.

Keratin 12 is a protein that in humans is encoded by the KRT12 gene. It's a type I keratin.

Recurrent corneal erosion is a disorder of the eyes characterized by the failure of the cornea's outermost layer of epithelial cells to attach to the underlying basement membrane. The condition is excruciatingly painful because the loss of these cells results in the exposure of sensitive corneal nerves. This condition can often leave patients with temporary blindness due to extreme light sensitivity (photophobia).

Decorin is a protein that in humans is encoded by the DCN gene.

Collagen XVII, previously called BP180, is a transmembrane protein which plays a critical role in maintaining the linkage between the intracellular and the extracellular structural elements involved in epidermal adhesion, identified by Diaz and colleagues in 1990.
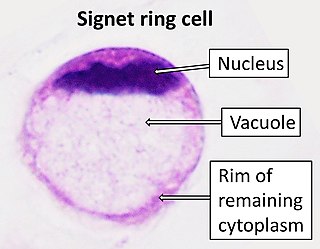
Signet ring cell carcinoma (SRCC) is a rare form of highly malignant adenocarcinoma that produces mucin. It is an epithelial malignancy characterized by the histologic appearance of signet ring cells.

Serine/threonine kinase 11 (STK11) also known as liver kinase B1 (LKB1) or renal carcinoma antigen NY-REN-19 is a protein kinase that in humans is encoded by the STK11 gene.

Transforming growth factor, beta-induced, 68kDa, also known as TGFBI, is a protein which in humans is encoded by the TGFBI gene, locus 5q31.

Peripherin-2 is a protein, that in humans is encoded by the PRPH2 gene. Peripherin-2 is found in the rod and cone cells of the retina of the eye. Defects in this protein result in one form of retinitis pigmentosa, an incurable blindness.
Serpin B3 is a protein that in humans is encoded by the SERPINB3 gene.
Zinc finger E-box-binding homeobox 1 is a protein that in humans is encoded by the ZEB1 gene.

Cone-rod homeobox protein is a protein that in humans is encoded by the CRX gene.

11-cis retinol dehydrogenase is an enzyme that in humans is encoded by the RDH5 gene.

Collagen alpha-2(VIII) chain is a protein that in humans is encoded by the COL8A2 gene. Mutations of the gene are linked to posterior polymorphous dystrophy type 2.

Sodium bicarbonate transporter-like protein 11 is a protein that in humans is encoded by the SLC4A11 gene.

Atypical fibroxanthoma (AFX) of the skin is a low-grade malignancy related to malignant fibrous histiocytoma, which it resembles histologically. Atypical fibroxanthoma manifests as a hard, pink or red papule or nodule that grows over the course of several months and may bleed or ulcerate. They typically occur on the head and neck. Atypical fibroxanthoma is usually asymptomatic.

Meesmann corneal dystrophy (MECD) is a rare hereditary autosomal dominant disease that is characterized as a type of corneal dystrophy and a keratin disease. MECD is characterized by the formation of microcysts in the outermost layer of the cornea, known as the anterior corneal epithelium. The anterior corneal epithelium also becomes fragile. This usually affects both eyes rather than a single eye and worsens over time. There are two phenotypes, Meesmann corneal dystrophy 1 (MECD1) and Meesmann corneal dystrophy 2 (MECD2), which affect the genes KRT3 and KRT12, respectively. A heterozygous mutation in either of these genes will lead to a single phenotype. Many with Meesmann corneal dystrophy are asymptomatic or experience mild symptoms.

Lisch epithelial corneal dystrophy (LECD), also known as band-shaped and whorled microcystic dystrophy of the corneal epithelium, is a rare form of corneal dystrophy first described in 1992 by Lisch et al. In one study it was linked to chromosomal region Xp22.3, with as yet unknown candidate genes.

Gelatinous drop-like corneal dystrophy, also known as amyloid corneal dystrophy, is a rare form of corneal dystrophy. The disease was described by Nakaizumi as early as 1914.
Occult macular dystrophy (OMD) is a rare inherited degradation of the retina, characterized by progressive loss of function in the most sensitive part of the central retina (macula), the location of the highest concentration of light-sensitive cells (photoreceptors) but presenting no visible abnormality. "Occult" refers to the degradation in the fundus being difficult to discern. The disorder is called "dystrophy" instead of "degradation" to distinguish its genetic origin from other causes, such as age. OMD was first reported by Y. Miyake et al. in 1989.






