Related Research Articles

Proteomics is the large-scale study of proteins. Proteins are vital macromolecules of all living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
PEAKS is a proteomics software program for tandem mass spectrometry designed for peptide sequencing, protein identification and quantification.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
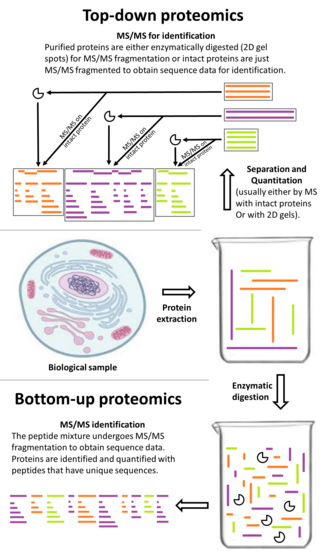
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
Label-free quantification is a method in mass spectrometry that aims to determine the relative amount of proteins in two or more biological samples. Unlike other methods for protein quantification, label-free quantification does not use a stable isotope containing compound to chemically bind to and thus label the protein.
An isotope-coded affinity tag (ICAT) is an in-vitro isotopic labeling method used for quantitative proteomics by mass spectrometry that uses chemical labeling reagents. These chemical probes consist of three elements: a reactive group for labeling an amino acid side chain, an isotopically coded linker, and a tag for the affinity isolation of labeled proteins/peptides. The samples are combined and then separated through chromatography, then sent through a mass spectrometer to determine the mass-to-charge ratio between the proteins. Only cysteine containing peptides can be analysed. Since only cysteine containing peptides are analysed, often the post translational modification is lost.
OpenMS is an open-source project for data analysis and processing in mass spectrometry and is released under the 3-clause BSD licence. It supports most common operating systems including Microsoft Windows, MacOS and Linux.

Selected reaction monitoring (SRM), also called multiple reaction monitoring (MRM), is a method used in tandem mass spectrometry in which an ion of a particular mass is selected in the first stage of a tandem mass spectrometer and an ion product of a fragmentation reaction of the precursor ions is selected in the second mass spectrometer stage for detection.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with chemical groups that have nominally identical mass (isobaric), but vary in terms of distribution of heavy isotopes in their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy collision-induced dissociation (HCD) during tandem mass spectrometry yielding reporter ions of different masses.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
Chemoproteomics entails a broad array of techniques used to identify and interrogate protein-small molecule interactions. Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery, in which lead compounds are designed to interact with predetermined disease-driving biological targets. As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action. Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.
Renã A. S. Robinson is an associate professor and the Dorothy J. Wingfield Phillips Chancellor's Faculty Fellow in the department of chemistry at the Vanderbilt University, where she is the principal investigator of the RASR Laboratory.
Skyline is an open source software for targeted proteomics and metabolomics data analysis. It runs on Microsoft Windows and supports the raw data formats from multiple mass spectrometric vendors. It contains a graphical user interface to display chromatographic data for individual peptide or small molecule analytes.

Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.
References
- ↑ O'Brien JJ, O'Connell JD, Paulo JA, Thakurta S, Rose CM, Weekes MP, et al. (January 2018). "Compositional Proteomics: Effects of Spatial Constraints on Protein Quantification Utilizing Isobaric Tags". Journal of Proteome Research. 17 (1): 590–599. doi:10.1021/acs.jproteome.7b00699. PMC 5806995 . PMID 29195270.
- ↑ Brenes A, Hukelmann J, Bensaddek D, Lamond AI (October 2019). "Multibatch TMT Reveals False Positives, Batch Effects and Missing Values". Molecular & Cellular Proteomics. 18 (10): 1967–1980. doi: 10.1074/mcp.RA119.001472 . PMC 6773557 . PMID 31332098.
- ↑ O'Connell JD, Paulo JA, O'Brien JJ, Gygi SP (May 2018). "Proteome-Wide Evaluation of Two Common Protein Quantification Methods". Journal of Proteome Research. 17 (5): 1934–1942. doi:10.1021/acs.jproteome.8b00016. PMC 5984592 . PMID 29635916.
- ↑ Tsai CF, Smith JS, Krajewski K, Zhao R, Moghieb AM, Nicora CD, et al. (September 2019). "Tandem Mass Tag Labeling Facilitates Reversed-Phase Liquid Chromatography-Mass Spectrometry Analysis of Hydrophilic Phosphopeptides". Analytical Chemistry. 91 (18): 11606–11613. doi:10.1021/acs.analchem.9b01814. PMC 7197904 . PMID 31418558.
- ↑ Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, et al. (April 2003). "Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS". Analytical Chemistry. 75 (8): 1895–904. doi:10.1021/ac0262560. PMID 12713048.
- ↑ Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, et al. (April 2008). "Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags". Analytical Chemistry. 80 (8): 2921–31. doi:10.1021/ac702422x. PMID 18312001.
- ↑ Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M, Savitski MM (April 2014). "Ion coalescence of neutron encoded TMT 10-plex reporter ions". Analytical Chemistry. 86 (7): 3594–601. doi:10.1021/ac500140s. PMID 24579773.
- ↑ Peshkin, L.; Ryazanova, L.; Wuhr, M.; et al. (2017). "Bayesian Confidence Intervals for Multiplexed Proteomics Integrate Ion-Statistics with Peptide Quantification Concordance". bioRxiv 10.1101/210476 .
- ↑ Paulo JA, O'Connell JD, Gygi SP (October 2016). "A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments". Journal of the American Society for Mass Spectrometry. 27 (10): 1620–5. Bibcode:2016JASMS..27.1620P. doi:10.1007/s13361-016-1434-9. PMC 5018445 . PMID 27400695.
- ↑ Specht H, Slavov N (January 2021). "Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers". Journal of Proteome Research. 20 (1): 880–887. doi:10.1021/acs.jproteome.0c00675. PMC 7775882 . PMID 33190502.
- ↑ Budnik B, Levy E, Harmange G, Slavov N (October 2018). "SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation". Genome Biology. 19 (1): 161. doi: 10.1186/s13059-018-1547-5 . PMC 6196420 . PMID 30343672.
- ↑ Slavov N (February 2021). "Single-cell protein analysis by mass spectrometry". Current Opinion in Chemical Biology. 60: 1–9. arXiv: 2004.02069 . doi:10.1016/j.cbpa.2020.04.018. PMC 7767890 . PMID 32599342.