
Inductively coupled plasma mass spectrometry (ICP-MS) is a type of mass spectrometry that uses an inductively coupled plasma to ionize the sample. It atomizes the sample and creates atomic and small polyatomic ions, which are then detected. It is known and used for its ability to detect metals and several non-metals in liquid samples at very low concentrations. It can detect different isotopes of the same element, which makes it a versatile tool in isotopic labeling.

Proteomics is the large-scale study of proteins. Proteins are vital macromolecules of all living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.

Stable isotope labeling by/with amino acids in cell culture (SILAC) is a technique based on mass spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling. It is a popular method for quantitative proteomics.
Surface-enhanced laser desorption/ionization (SELDI) is a soft ionization method in mass spectrometry (MS) used for the analysis of protein mixtures. It is a variation of matrix-assisted laser desorption/ionization (MALDI). In MALDI, the sample is mixed with a matrix material and applied to a metal plate before irradiation by a laser, whereas in SELDI, proteins of interest in a sample become bound to a surface before MS analysis. The sample surface is a key component in the purification, desorption, and ionization of the sample. SELDI is typically used with time-of-flight (TOF) mass spectrometers and is used to detect proteins in tissue samples, blood, urine, or other clinical samples, however, SELDI technology can potentially be used in any application by simply modifying the sample surface.
Phosphoproteomics is a branch of proteomics that identifies, catalogs, and characterizes proteins containing a phosphate group as a posttranslational modification. Phosphorylation is a key reversible modification that regulates protein function, subcellular localization, complex formation, degradation of proteins and therefore cell signaling networks. With all of these modification results, it is estimated that between 30–65% of all proteins may be phosphorylated, some multiple times. Based on statistical estimates from many datasets, 230,000, 156,000 and 40,000 phosphorylation sites should exist in human, mouse, and yeast, respectively.
In analytical chemistry, a tandem mass tag (TMT) is a chemical label that facilitates sample multiplexing in mass spectrometry (MS)-based quantification and identification of biological macromolecules such as proteins, peptides and nucleic acids. TMT belongs to a family of reagents referred to as isobaric mass tags which are a set of molecules with the same mass, but yield reporter ions of differing mass after fragmentation. The relative ratio of the measured reporter ions represents the relative abundance of the tagged molecule, although ion suppression has a detrimental effect on accuracy. Despite these complications, TMT-based proteomics has been shown to afford higher precision than label-free quantification. In addition to aiding in protein quantification, TMT tags can also increase the detection sensitivity of certain highly hydrophilic analytes, such as phosphopeptides, in RPLC-MS analyses.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
An isotope-coded affinity tag (ICAT) is an in-vitro isotopic labeling method used for quantitative proteomics by mass spectrometry that uses chemical labeling reagents. These chemical probes consist of three elements: a reactive group for labeling an amino acid side chain, an isotopically coded linker, and a tag for the affinity isolation of labeled proteins/peptides. The samples are combined and then separated through chromatography, then sent through a mass spectrometer to determine the mass-to-charge ratio between the proteins. Only cysteine containing peptides can be analysed. Since only cysteine containing peptides are analysed, often the post translational modification is lost.
OpenMS is an open-source project for data analysis and processing in mass spectrometry and is released under the 3-clause BSD licence. It supports most common operating systems including Microsoft Windows, MacOS and Linux.

Selected reaction monitoring (SRM), also called multiple reaction monitoring (MRM), is a method used in tandem mass spectrometry in which an ion of a particular mass is selected in the first stage of a tandem mass spectrometer and an ion product of a fragmentation reaction of the precursor ions is selected in the second mass spectrometer stage for detection.
Bioanalysis is a sub-discipline of analytical chemistry covering the quantitative measurement of xenobiotics and biotics in biological systems.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with chemical groups that have nominally identical mass (isobaric), but vary in terms of distribution of heavy isotopes in their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy collision-induced dissociation (HCD) during tandem mass spectrometry yielding reporter ions of different masses.
A peptide spectral library is a curated, annotated and non-redundant collection/database of LC-MS/MS peptide spectra. One essential utility of a peptide spectral library is to serve as consensus templates supporting the identification of peptides and proteins based on the correlation between the templates with experimental spectra.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.
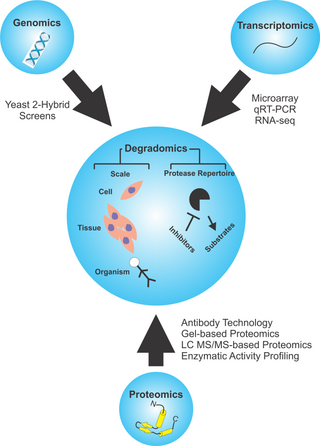
Degradomics is a sub-discipline of biology encompassing all the genomic and proteomic approaches devoted to the study of proteases, their inhibitors, and their substrates on a system-wide scale. This includes the analysis of the protease and protease-substrate repertoires, also called "protease degradomes". The scope of these degradomes can range from cell, tissue, and organism-wide scales.
Stable isotope standards and capture by anti-peptide antibodies (SISCAPA) is a mass spectrometry method for measuring the amount of a protein in a biological sample.
