
Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
Protein sequencing is the practical process of determining the amino acid sequence of all or part of a protein or peptide. This may serve to identify the protein or characterize its post-translational modifications. Typically, partial sequencing of a protein provides sufficient information to identify it with reference to databases of protein sequences derived from the conceptual translation of genes.

Electron-capture dissociation (ECD) is a method of fragmenting gas-phase ions for structure elucidation of peptides and proteins in tandem mass spectrometry. It is one of the most widely used techniques for activation and dissociation of mass selected precursor ion in MS/MS. It involves the direct introduction of low-energy electrons to trapped gas-phase ions.
Hydrogen–deuterium exchange is a chemical reaction in which a covalently bonded hydrogen atom is replaced by a deuterium atom, or vice versa. It can be applied most easily to exchangeable protons and deuterons, where such a transformation occurs in the presence of a suitable deuterium source, without any catalyst. The use of acid, base or metal catalysts, coupled with conditions of increased temperature and pressure, can facilitate the exchange of non-exchangeable hydrogen atoms, so long as the substrate is robust to the conditions and reagents employed. This often results in perdeuteration: hydrogen-deuterium exchange of all non-exchangeable hydrogen atoms in a molecule.

Stable Isotope Labeling by/with Amino acids in Cell culture (SILAC) is a technique based on mass spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling. It is a popular method for quantitative proteomics.
A peptide sequence tag is a piece of information about a peptide obtained by tandem mass spectrometry that can be used to identify this peptide in a protein database.

Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.
A tandem mass tag (TMT) is a chemical label that facilitates sample multiplexing in mass spectrometry (MS)-based quantification and identification of biological macromolecules such as proteins, peptides and nucleic acids. TMT belongs to a family of reagents referred to as isobaric mass tags which are a set of molecules with the same mass, but yield reporter ions of differing mass after fragmentation. The relative ratio of the measured reporter ions represents the relative abundance of the tagged molecule, although ion suppression has a detrimental effect on accuracy. Despite these complications, TMT-based proteomics has been shown to afford higher precision than Label-free quantification. In addition to aiding in protein quantification, TMT tags can also increase the detection sensitivity of certain highly hydrophilic analytes, such as phosphopeptides, in RPLC-MS analyses.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
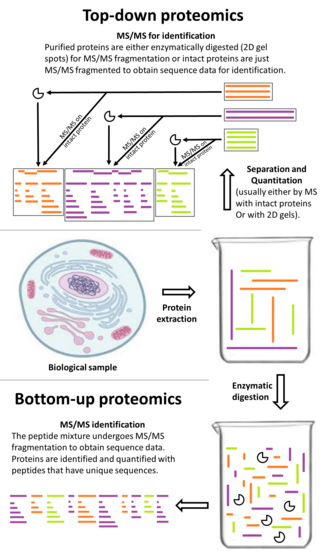
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.
Label-free quantification is a method in mass spectrometry that aims to determine the relative amount of proteins in two or more biological samples. Unlike other methods for protein quantification, label-free quantification does not use a stable isotope containing compound to chemically bind to and thus label the protein.
An Isotope-coded affinity tag (ICAT) is an in-vitro isotopic labeling method used for quantitative proteomics by mass spectrometry that uses chemical labeling reagents. These chemical probes consist of three elements: a reactive group for labeling an amino acid side chain, an isotopically coded linker, and a tag for the affinity isolation of labeled proteins/peptides. The samples are combined and then separated through chromatography, then sent through a mass spectrometer to determine the mass-to-charge ratio between the proteins. Only cysteine containing peptides can be analysed. Since only cysteine containing peptides are analysed, often the post translational modification is lost.

Selected reaction monitoring (SRM), also called Multiple reaction monitoring, (MRM), is a method used in tandem mass spectrometry in which an ion of a particular mass is selected in the first stage of a tandem mass spectrometer and an ion product of a fragmentation reaction of the precursor ions is selected in the second mass spectrometer stage for detection.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with chemical groups that have identical mass (isobaric), but vary in terms of distribution of heavy isotopes in their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy CID (HCD) during tandem mass spectrometry yielding reporter ions of different masses. The most common isobaric tags are amine-reactive tags. However, tags that react with cysteine residues and carbonyl groups have also been described. These amine-reactive groups go through N-hydroxysuccinimide (NHS) reactions, which are based around three types of functional groups. Isobaric labeling methods include tandem mass tags (TMT), isobaric tags for relative and absolute quantification (iTRAQ), mass differential tags for absolute and relative quantification, and dimethyl labeling. TMTs and iTRAQ methods are most common and developed of these methods. Tandem mass tags have a mass reporter region, a cleavable linker region, a mass normalization region, and a protein reactive group and have the same total mass.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.
Renã A. S. Robinson is an associate professor and the Dorothy J. Wingfield Phillips Chancellor's Faculty Fellow in the department of chemistry at the Vanderbilt University, where she is the principal investigator of the RASR Laboratory.
Skyline is an open source software for targeted proteomics and metabolomics data analysis. It runs on Microsoft Windows and supports the raw data formats from multiple mass spectrometric vendors. It contains a graphical user interface to display chromatographic data for individual peptide or small molecule analytes.
