
A protein kinase is a kinase which selectively modifies other proteins by covalently adding phosphates to them (phosphorylation) as opposed to kinases which modify lipids, carbohydrates, or other molecules. Phosphorylation usually results in a functional change of the target protein (substrate) by changing enzyme activity, cellular location, or association with other proteins. The human genome contains about 500 protein kinase genes and they constitute about 2% of all human genes. There are two main types of protein kinase. The great majority are serine/threonine kinases, which phosphorylate the hydroxyl groups of serines and threonines in their targets and most of the others are tyrosine kinases, although additional types exist. Protein kinases are also found in bacteria and plants. Up to 30% of all human proteins may be modified by kinase activity, and kinases are known to regulate the majority of cellular pathways, especially those involved in signal transduction.
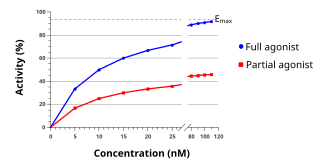
An agonist is a chemical that activates a receptor to produce a biological response. Receptors are cellular proteins whose activation causes the cell to modify what it is currently doing. In contrast, an antagonist blocks the action of the agonist, while an inverse agonist causes an action opposite to that of the agonist.

A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. Antagonist drugs interfere in the natural operation of receptor proteins. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

Gefitinib, sold under the brand name Iressa, is a medication used for certain breast, lung and other cancers. Gefitinib is an EGFR inhibitor, like erlotinib, which interrupts signaling through the epidermal growth factor receptor (EGFR) in target cells. Therefore, it is only effective in cancers with mutated and overactive EGFR, but resistances to gefitinib can arise through other mutations. It is marketed by AstraZeneca and Teva.

The epidermal growth factor receptor is a transmembrane protein that is a receptor for members of the epidermal growth factor family of extracellular protein ligands.
Quinazoline is an organic compound with the formula C8H6N2. It is an aromatic heterocycle with a bicyclic structure consisting of two fused six-membered aromatic rings, a benzene ring and a pyrimidine ring. It is a light yellow crystalline solid that is soluble in water. Also known as 1,3-diazanaphthalene, quinazoline received its name from being an aza derivative of quinoline. Though the parent quinazoline molecule is rarely mentioned by itself in technical literature, substituted derivatives have been synthesized for medicinal purposes such as antimalarial and anticancer agents. Quinazoline is a planar molecule. It is isomeric with the other diazanaphthalenes of the benzodiazine subgroup: cinnoline, quinoxaline, and phthalazine. Over 200 biologically active quinazoline and quinoline alkaloids are identified.

Erlotinib, sold under the brand name Tarceva among others, is a medication used to treat non-small cell lung cancer (NSCLC) and pancreatic cancer. Specifically it is used for NSCLC with mutations in the epidermal growth factor receptor (EGFR) — either an exon 19 deletion (del19) or exon 21 (L858R) substitution mutation — which has spread to other parts of the body. It is taken by mouth.
Affinity labels are a class of enzyme inhibitors that covalently bind to their target causing its inactivation. The hallmark of an affinity label is the use of a targeting moiety to specifically and reversibly deliver a weakly reactive group to the enzyme that irreversibly binds to an amino acid residue. The targeting portion of the label often resembles the enzyme's natural substrate so that a similar mode of noncovalent binding is used prior to the covalent linkage. Their usefulness in medicine can be limited by the specificity of the first noncovalent binding step whereas indiscriminate action can be utilized for purposes such as affinity labeling - a technique for the validation of substrate-specific binding of compounds.

An enzyme inhibitor is a molecule that binds to an enzyme and blocks its activity. Enzymes are proteins that speed up chemical reactions necessary for life, in which substrate molecules are converted into products. An enzyme facilitates a specific chemical reaction by binding the substrate to its active site, a specialized area on the enzyme that accelerates the most difficult step of the reaction.

KRAS is a gene that provides instructions for making a protein called K-Ras, a part of the RAS/MAPK pathway. The protein relays signals from outside the cell to the cell's nucleus. These signals instruct the cell to grow and divide (proliferate) or to mature and take on specialized functions (differentiate). It is called KRAS because it was first identified as a viral oncogene in the KirstenRAt Sarcoma virus. The oncogene identified was derived from a cellular genome, so KRAS, when found in a cellular genome, is called a proto-oncogene.
A protein kinase inhibitor is a type of enzyme inhibitor that blocks the action of one or more protein kinases. Protein kinases are enzymes that phosphorylate (add a phosphate, or PO4, group) to a protein and can modulate its function.

Staurosporine is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus. It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.
Bioconjugation is a chemical strategy to form a stable covalent link between two molecules, at least one of which is a biomolecule.
Molecular binding is an attractive interaction between two molecules that results in a stable association in which the molecules are in close proximity to each other. It is formed when atoms or molecules bind together by sharing of electrons. It often, but not always, involves some chemical bonding.

A tyrosine kinase inhibitor (TKI) is a pharmaceutical drug that inhibits tyrosine kinases. Tyrosine kinases are enzymes responsible for the activation of many proteins by signal transduction cascades. The proteins are activated by adding a phosphate group to the protein (phosphorylation), a step that TKIs inhibit. TKIs are typically used as anticancer drugs. For example, they have substantially improved outcomes in chronic myelogenous leukemia. They have also been used to treat other diseases, such as idiopathic pulmonary fibrosis.
Bcr-Abl tyrosine-kinase inhibitors (TKI) are the first-line therapy for most patients with chronic myelogenous leukemia (CML). More than 90% of CML cases are caused by a chromosomal abnormality that results in the formation of a so-called Philadelphia chromosome. This abnormality was discovered by Peter Nowell in 1960 and is a consequence of fusion between the Abelson (Abl) tyrosine kinase gene at chromosome 9 and the break point cluster (Bcr) gene at chromosome 22, resulting in a chimeric oncogene (Bcr-Abl) and a constitutively active Bcr-Abl tyrosine kinase that has been implicated in the pathogenesis of CML. Compounds have been developed to selectively inhibit the tyrosine kinase.
c-Met inhibitors are a class of small molecules that inhibit the enzymatic activity of the c-Met tyrosine kinase, the receptor of hepatocyte growth factor/scatter factor (HGF/SF). These inhibitors may have therapeutic application in the treatment of various types of cancers.

Osimertinib, sold under the brand name Tagrisso, is a medication used to treat non-small-cell lung carcinomas with specific mutations. It is a third-generation epidermal growth factor receptor tyrosine kinase inhibitor.

Olmutinib (INN) is an investigational anti-cancer drug. It acts by covalently bonding to a cysteine residue near the kinase domain of epidermal growth factor receptor (EGFR).

Mobocertinib, sold under the brand name Exkivity, is used for the treatment of non-small cell lung cancer.



