
A mitochondrion is an organelle found in the cells of most eukaryotes, such as animals, plants and fungi. Mitochondria have a double membrane structure and use aerobic respiration to generate adenosine triphosphate (ATP), which is used throughout the cell as a source of chemical energy. They were discovered by Albert von Kölliker in 1857 in the voluntary muscles of insects. The term mitochondrion was coined by Carl Benda in 1898. The mitochondrion is popularly nicknamed the "powerhouse of the cell", a phrase coined by Philip Siekevitz in a 1957 article of the same name.
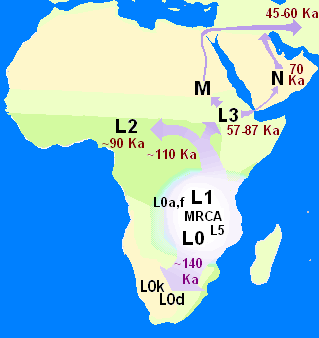
In human genetics, the Mitochondrial Eve is the matrilineal most recent common ancestor (MRCA) of all living humans. In other words, she is defined as the most recent woman from whom all living humans descend in an unbroken line purely through their mothers and through the mothers of those mothers, back until all lines converge on one woman.

Mitochondrial DNA is the DNA located in mitochondria, cellular organelles within eukaryotic cells that convert chemical energy from food into a form that cells can use, such as adenosine triphosphate (ATP). Mitochondrial DNA is only a small portion of the DNA in a eukaryotic cell; most of the DNA can be found in the cell nucleus and, in plants and algae, also in plastids such as chloroplasts.

An electron transport chain (ETC) is a series of protein complexes and other molecules that transfer electrons from electron donors to electron acceptors via redox reactions (both reduction and oxidation occurring simultaneously) and couples this electron transfer with the transfer of protons (H+ ions) across a membrane. The electrons that are transferred from NADH and FADH2 to the ETC involves four multi-subunit large enzymes complexes and two mobile electron carriers. Many of the enzymes in the electron transport chain are membrane-bound.

Mitochondrial disease is a group of disorders caused by mitochondrial dysfunction. Mitochondria are the organelles that generate energy for the cell and are found in every cell of the human body except red blood cells. They convert the energy of food molecules into the ATP that powers most cell functions.
Creatine kinase (CK), also known as creatine phosphokinase (CPK) or phosphocreatine kinase, is an enzyme expressed by various tissues and cell types. CK catalyses the conversion of creatine and uses adenosine triphosphate (ATP) to create phosphocreatine (PCr) and adenosine diphosphate (ADP). This CK enzyme reaction is reversible and thus ATP can be generated from PCr and ADP.

Human mitochondrial genetics is the study of the genetics of human mitochondrial DNA. The human mitochondrial genome is the entirety of hereditary information contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.
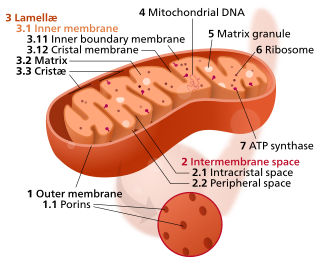
The inner mitochondrial membrane (IMM) is the mitochondrial membrane which separates the mitochondrial matrix from the intermembrane space.

In human genetics, a human mitochondrial DNA haplogroup is a haplogroup defined by differences in human mitochondrial DNA. Haplogroups are used to represent the major branch points on the mitochondrial phylogenetic tree. Understanding the evolutionary path of the female lineage has helped population geneticists trace the matrilineal inheritance of modern humans back to human origins in Africa and the subsequent spread around the globe.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.

The MRC Mitochondrial Biology Unit is a department of the School of Clinical Medicine at the University of Cambridge, funded through a strategic partnership between the Medical Research Council and the University. It is located at the Addenbrooke’s Hospital / Cambridge Biomedical Campus site in Cambridge, England. The unit is concerned with the study of the mitochondrion, as this organelle has a varied and critical role in many aspects of eukaryotic metabolism and is implicated in many metabolic, degenerative, and age-related human diseases.
Mitochondrial carriers are proteins from solute carrier family 25 which transfer molecules across the membranes of the mitochondria. Mitochondrial carriers are also classified in the Transporter Classification Database. The Mitochondrial Carrier (MC) Superfamily has been expanded to include both the original Mitochondrial Carrier (MC) family and the Mitochondrial Inner/Outer Membrane Fusion (MMF) family.

Nuclear respiratory factor 1, also known as Nrf1, Nrf-1, NRF1 and NRF-1, encodes a protein that homodimerizes and functions as a transcription factor which activates the expression of some key metabolic genes regulating cellular growth and nuclear genes required for respiration, heme biosynthesis, and mitochondrial DNA transcription and replication. The protein has also been associated with the regulation of neurite outgrowth. Alternate transcriptional splice variants, which encode the same protein, have been characterized. Additional variants encoding different protein isoforms have been described but they have not been fully characterized. Confusion has occurred in bibliographic databases due to the shared symbol of NRF1 for this gene and for "nuclear factor -like 1" which has an official symbol of NFE2L1.

DNA polymerase subunit gamma is an enzyme that in humans is encoded by the POLG gene. Mitochondrial DNA polymerase is heterotrimeric, consisting of a homodimer of accessory subunits plus a catalytic subunit. The protein encoded by this gene is the catalytic subunit of mitochondrial DNA polymerase. Defects in this gene are a cause of progressive external ophthalmoplegia with mitochondrial DNA deletions 1 (PEOA1), sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO), Alpers-Huttenlocher syndrome (AHS), and mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE).

Dynamin-like 120 kDa protein, mitochondrial is a protein that in humans is encoded by the OPA1 gene. This protein regulates mitochondrial fusion and cristae structure in the inner mitochondrial membrane (IMM) and contributes to ATP synthesis and apoptosis, and small, round mitochondria. Mutations in this gene have been implicated in dominant optic atrophy (DOA), leading to loss in vision, hearing, muscle contraction, and related dysfunctions.

Brain mitochondrial carrier protein 1 is a protein that in humans is encoded by the SLC25A14 gene.

DNA-directed RNA polymerase, mitochondrial is an enzyme that in humans is encoded by the POLRMT gene.
Mitochondrially encoded tRNA valine also known as MT-TV is a transfer RNA which in humans is encoded by the mitochondrial MT-TV gene.

The mitochondrial ribosome, or mitoribosome, is a protein complex that is active in mitochondria and functions as a riboprotein for translating mitochondrial mRNAs encoded in mtDNA. The mitoribosome is attached to the inner mitochondrial membrane. Mitoribosomes, like cytoplasmic ribosomes, consist of two subunits — large (mt-LSU) and small (mt-SSU). Mitoribosomes consist of several specific proteins and fewer rRNAs. While mitochondrial rRNAs are encoded in the mitochondrial genome, the proteins that make up mitoribosomes are encoded in the nucleus and assembled by cytoplasmic ribosomes before being implanted into the mitochondria.
